nosoiSim object
The output of a nosoi simulation is an object of class
nosoiSim that contains all the data generated during the
simulation into the following slots:
-
total.timeis the number of time steps (an integer) during which the simulation has run. -
typeis the simulation type that was ran (either “single” or “dual” host). -
host.info.Ais an object of the classnosoiSimOne, itself a list of elements concerning host type A (or the unique host type in case of a single-host simulation), with the following slots:-
N.infectedis the final number (an integer) of infected hosts. -
table.hostsis a table containing the results of the simulations focusing on each individual host. For more details on its structure, see below. -
table.stateis a table containing the results of the simulations concerning the movement history of the infected hosts. For more details on its structure, see below. -
prefix.hostis the string containing the prefix used to name the hosts (user-definedprefix.hostin thenosoiSimfunction). -
popStructureis the population structure used (either “none”, “discrete” or “continuous”).
-
-
host.info.Bis the same as above, but for host type B (but is set toNAif a single-host was run).
Host-specific elements (i.e. in host.info.A or
host.info.B) in this object can be easily extracted using
the function getHostData:
getHostData(nosoi.output, what, pop)This function takes as its arguments your nosoi output object (of
class nosoiSim), what you want to extract
(either N.infected, table.hosts,
table.state or prefix.host), and which host
type (here pop) you are interested in (A by
default, but can be B in case of dual-host
simulations).
table.hosts
table.hosts, a data.table object, contains
information about each host that has been simulated. In this object, one
row corresponds to one host. This object can be directly extracted using
the function getTableHosts:
getTableHosts(nosoi.output, pop)This function takes as its arguments your nosoi output object (of
class nosoiSim) and which host type (here pop)
you are interested in (A by default but can be
B in case of dual-host simulations).
The structure of the table is the following:
-
hosts.IDis the unique identifier for the host (a string of characters), based on a user-defined prefix and an integer. -
inf.byis the unique identifier of the host that infected the current one (stands for “infected by”). -
inf.in(only if structure is present) is the (discrete) location (e.g. province, country, …) or coordinates (in that caseinf.in.xandinf.in.y) in which the host was initially infected (stands for “infected in”). -
current.in(only if structure is present) is the (discrete) location (e.g. province, country, …) or coordinates (in that casecurrent.in.xandcurrent.in.y) in which the host is at the end of the simulation (stands for “currently in”). A complete history of hosts movements is stored in the other tabletable.state(see following section). -
current.env.value(only if continuous structure is present) is the environmental value (i.e. raster cell value) in which the host is at the end of the simulation (stands for “current environmental value”). A complete history of the environmental value encountered by the host is stored in the other tabletable.state(see following section). -
current.cell.raster(only if continuous structure is present) is the raster cell numeric identifier in which the host is at the end of the simulation. Here also, a complete history of raster cell occupied during the simulation is stored in the other tabletable.state(see following section). -
host.count(only if structure is present) is the host count in the current state or raster cell. This value is only updated if the host count is actually used as a parameter in the simulation. -
inf.timeis the time (in time steps) at which the host entered the simulation (i.e. got infected) (stands for “infection time”). -
out.timeis the time (in time steps) at which the host exited the simulation. -
activeis a number (either 0 or 1) indicating if the host is still active at the end of the simulation (1 for yes, 0 for no). - the rest stores the user-defined individual-specific parameters used
by the functions (i.e. specified by the
paramarguments in the simulator’s functions).
table.state
table.state, a data.table object, contains
information about the history of movement of each host that has been
simulated. This table is available only if a structured population was
simulated (either discrete or continuous). It can be directly extracted
using the function getTableState:
getTableState(nosoi.output, pop)This function takes as its arguments your nosoi output object (of
class nosoiSim) and which host type (here pop)
you are interested in (A by default but can be
B in case of dual-host simulations).
The structure of the table is the following:
-
hosts.IDis the unique identifier for the host (a string of characters), based on a user-defined prefix and an integer. -
stateis the (discrete) location (e.g. province, country, …) or coordinates (in that casestate.xandstate.y) in which the host is located during the time interval. -
current.env.value(only if continuous structure is present) is the environmental value (i.e. raster cell value) in which the host is at the end of the simulation (stands for “current environmental value”). -
current.cell.raster(only if continuous structure is present) is the raster cell numeric identifier in which the host is at the end of the simulation. -
time.fromis the time (in time steps) at which the host entered (i.e. got infected or moved) into the state. -
time.tois the time (in time steps) at which the host left (i.e. excited or moved) out of the state.
Epidemiological insights
By simulating transmission chains, nosoi also simulates
an epidemic process. nosoi provides some solutions to
easily explore the dynamics of this epidemic process by following
through time the number of active infected hosts as well as the
cumulative number of infected hosts. It also allows to compute the exact
basic reproduction number \(R_0\),
defined as the average number of cases one case generates. Since all of
the data generated are stored in the output nosoiSim
object, more advanced exploration could also be done.
Epidemiological summary
summary is a turnkey solution that computes both the
dynamics of the epidemic and \(R_0\).
Its only argument is your nosoi output object (of class
nosoiSim):
summary(nosoi.output)It returns a list containing the slots:
-
R0, a sub-list with number of inactive hosts at the end of the simulationN.inactiveon which the calculation is done, mean \(R_0\)R0.mean, and \(R_0\) distributionR0.dist. For more details on the computation behind, see below. -
dynamicsis adata.tableobject with the count of the sum of infected and active host at each time of the simulation (by location/state if this simulation has been done in a discrete structured host population). See below for more details. -
cumulativeis adata.tableobject with the cumulative sum of infected hosts at each time step of the simulation. See below for more details.
Epidemiological dynamics
Both the cumulative and dynamics tables can
be directly extracted using the functions getCumulative and
getDynamic respectively. Both functions yield a
data.table object, but their structures vary slightly.
cumulative has the following structure:
-
tis the time step (an integer). -
Countis the cumulative number of infected hosts at given time step (an integer). -
typeis the host type, identified by its user-defined prefix (string of characters).
dynamics has the following structure:
-
state(only in the case of a discrete structure) is the state (a string of characters, usually). -
Countis the number of active infected hosts at given time step (an integer) by state (in the case of a discrete structure). -
typeis the host type, identified by its user-defined prefix (string of characters). -
tis the time step (an integer).
These can be used to plot the epidemiological dynamics of the simulated transmission chain. Here for example, we simulate a single introduction of a single-host pathogen between 3 different locations/states (named “A”, “B”, and “C”), with constant exit and move probabilities as well as a number of contacts dependent on the location and host count in each location:
library(nosoi)
t_incub_fct <- function(x){rnorm(x,mean = 5,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
p_Move_fct <- function(t){return(0.1)}
p_Exit_fct <- function(t){return(0.05)}
proba <- function(t,p_max,t_incub){
if(t <= t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
time_contact <- function(t, current.in, host.count){
temp.val = 30 - host.count
if(temp.val <= 0) {
return(0)
}
if(temp.val >= 0) {
if(current.in=="A"){
return(round((temp.val/30)*rnorm(1, 3, 1), 0))}
if(current.in=="B"){return(0)}
if(current.in=="C"){
return(round((temp.val/30)*rnorm(1, 6, 1), 0))}
}
}
transition.matrix = matrix(c(0,0.2,0.4,0.5,0,0.6,0.5,0.8,0),nrow = 3, ncol = 3,dimnames=list(c("A","B","C"),c("A","B","C")))
set.seed(1050)
test.nosoiA <- nosoiSim(type="single", popStructure="discrete",
length=100,
max.infected=200,
init.individuals=1,
init.structure="A",
structure.matrix=transition.matrix,
pMove=p_Move_fct,
param.pMove=NA,
diff.nContact=TRUE,
hostCount.nContact=TRUE,
nContact=time_contact,
param.nContact=NA,
pTrans = proba,
param.pTrans = list(p_max=p_max_fct,
t_incub=t_incub_fct),
pExit=p_Exit_fct,
param.pExit=NA
)
library(ggplot2)
cumulative.table <- getCumulative(test.nosoiA)
dynamics.table <- getDynamic(test.nosoiA)
ggplot(data=cumulative.table, aes(x=t, y=Count)) + geom_line() + theme_minimal() + labs(x="Time (t)",y="Cumulative count of infected hosts")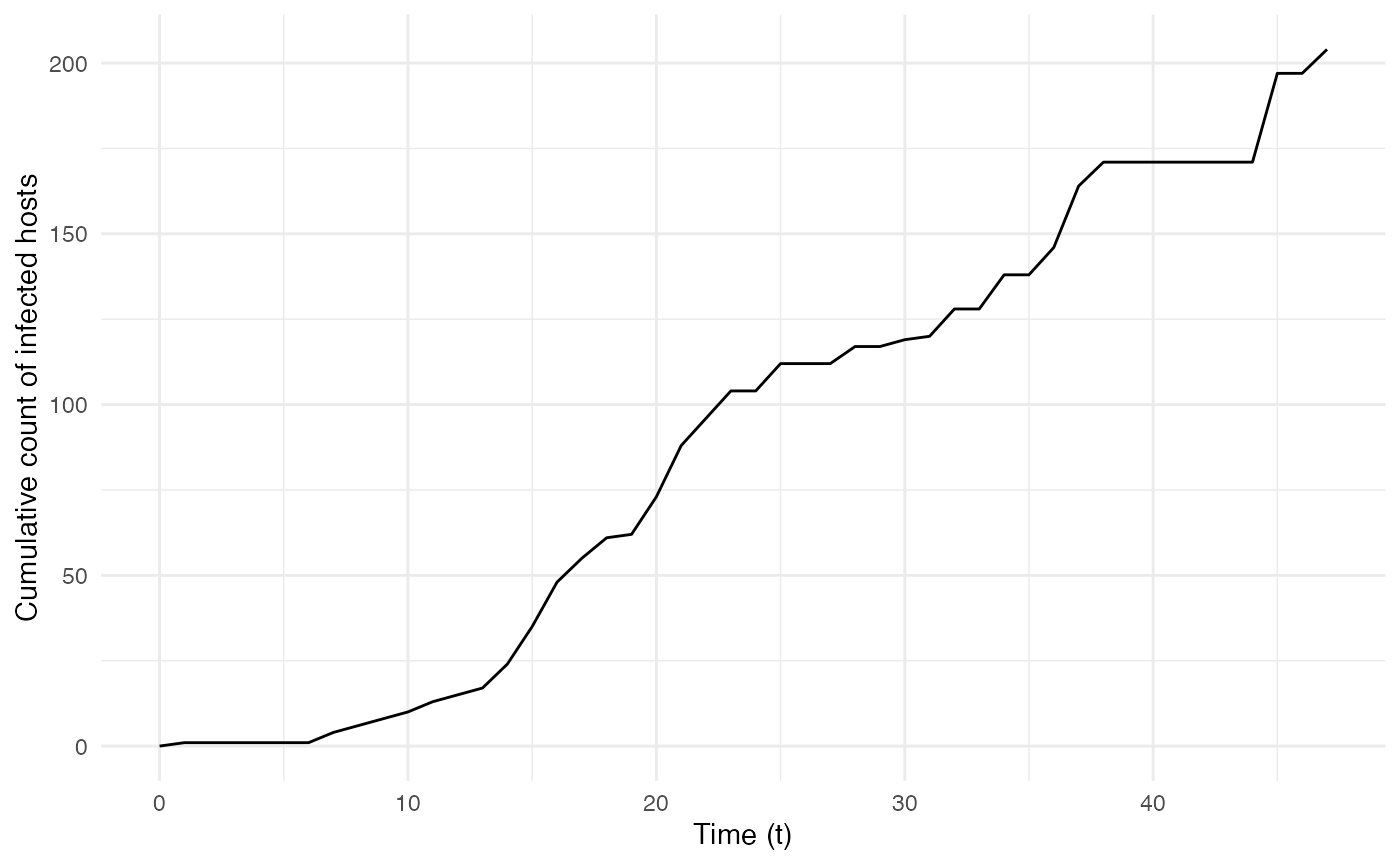
ggplot(data=dynamics.table, aes(x=t, y=Count, color=state)) + geom_line() + theme_minimal() + labs(x="Time (t)",y="Number of active infected hosts")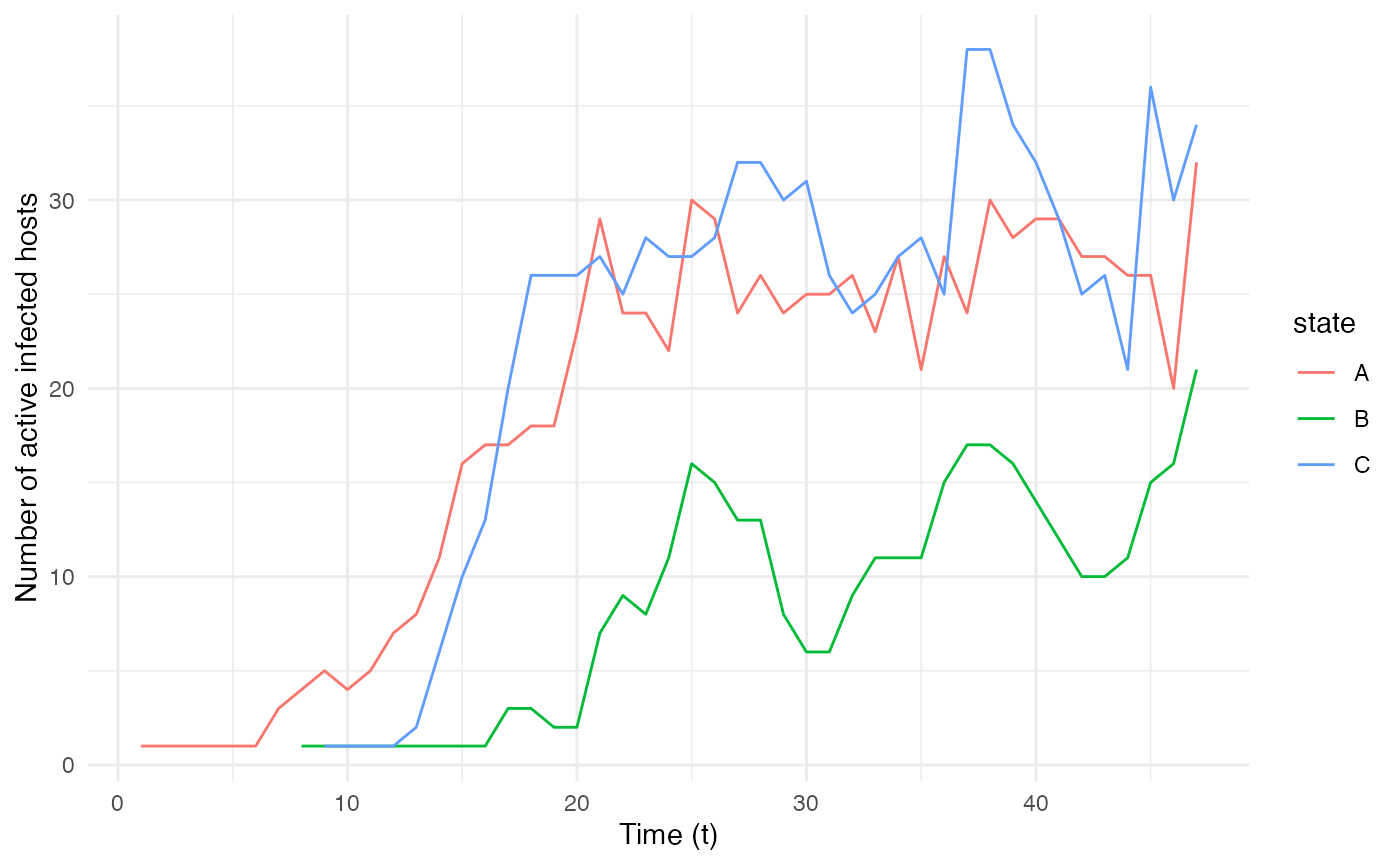
\(R_0\)
The output nosoiSim object can be used to compute the
“real” \(R_0\), defined as the average
number of cases one case generates, often estimated in epidemiological
studies. The function getRO can be used directly, with the
nosoiSim output as its unique argument, to generate a list
containing:
-
N.inactive, the number of inactive hosts at the end of the simulation on which the \(R_0\) calculation is made. This is to avoid bias introduced by hosts that have not accomplished their full transmission potential (i.e. are still active). -
R0.meanis the \(R_0\) estimate, mean of the followingR0.dist. -
R0.distis the full distribution (one number is one host) of \(R_0\) estimates per host.
Here for example, we simulate a single introduction of a single-host pathogen between 3 different states (named “A”, “B”, and “C”), with a constant exit and move probability as well as a number of contacts dependent of the location and host count in each location:
library(nosoi)
t_incub_fct <- function(x){rnorm(x,mean = 5,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
p_Move_fct <- function(t){return(0.1)}
p_Exit_fct <- function(t){return(0.05)}
proba <- function(t,p_max,t_incub){
if(t <= t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
time_contact <- function(t, current.in, host.count){
temp.val = 30 - host.count
if(temp.val <= 0) {
return(0)
}
if(temp.val >= 0) {
if(current.in=="A"){
return(round((temp.val/30)*rnorm(1, 3, 1), 0))}
if(current.in=="B"){return(0)}
if(current.in=="C"){
return(round((temp.val/30)*rnorm(1, 6, 1), 0))}
}
}
transition.matrix = matrix(c(0,0.2,0.4,0.5,0,0.6,0.5,0.8,0),nrow = 3, ncol = 3,dimnames=list(c("A","B","C"),c("A","B","C")))
set.seed(1050)
test.nosoiA <- nosoiSim(type="single", popStructure="discrete",
length=100,
max.infected=200,
init.individuals=1,
init.structure="A",
structure.matrix=transition.matrix,
pMove=p_Move_fct,
param.pMove=NA,
diff.nContact=TRUE,
hostCount.nContact=TRUE,
nContact=time_contact,
param.nContact=NA,
pTrans = proba,
param.pTrans = list(p_max=p_max_fct,
t_incub=t_incub_fct),
pExit=p_Exit_fct,
param.pExit=NA
)
getR0(test.nosoiA)
#> $N.inactive
#> [1] 117
#>
#> $R0.mean
#> [1] 1.068376
#>
#> $R0.dist
#> [1] 18 4 0 0 0 0 0 0 6 0 0 0 0 0 0 0 0 0 0 6 0 0 0 0 0
#> [26] 4 0 0 0 0 9 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 3 10 0 2
#> [51] 1 4 5 0 0 0 1 2 1 0 3 2 0 0 0 11 0 0 0 0 1 0 8 0 1
#> [76] 0 1 1 0 0 0 1 0 0 0 0 0 0 0 1 2 0 3 0 0 1 0 3 0 3
#> [101] 0 0 0 0 0 1 0 1 0 0 1 0 1 2 1 0 0As you can see, out of the 117 inactive hosts, the mean \(R_0\) is 1.068376. This does not of course reflect the distribution of \(R_0\) where most hosts actually never transmitted the infection:
data = data.frame(R0=getR0(test.nosoiA)$R0.dist)
ggplot(data=data, aes(x=R0)) + geom_histogram() + theme_minimal()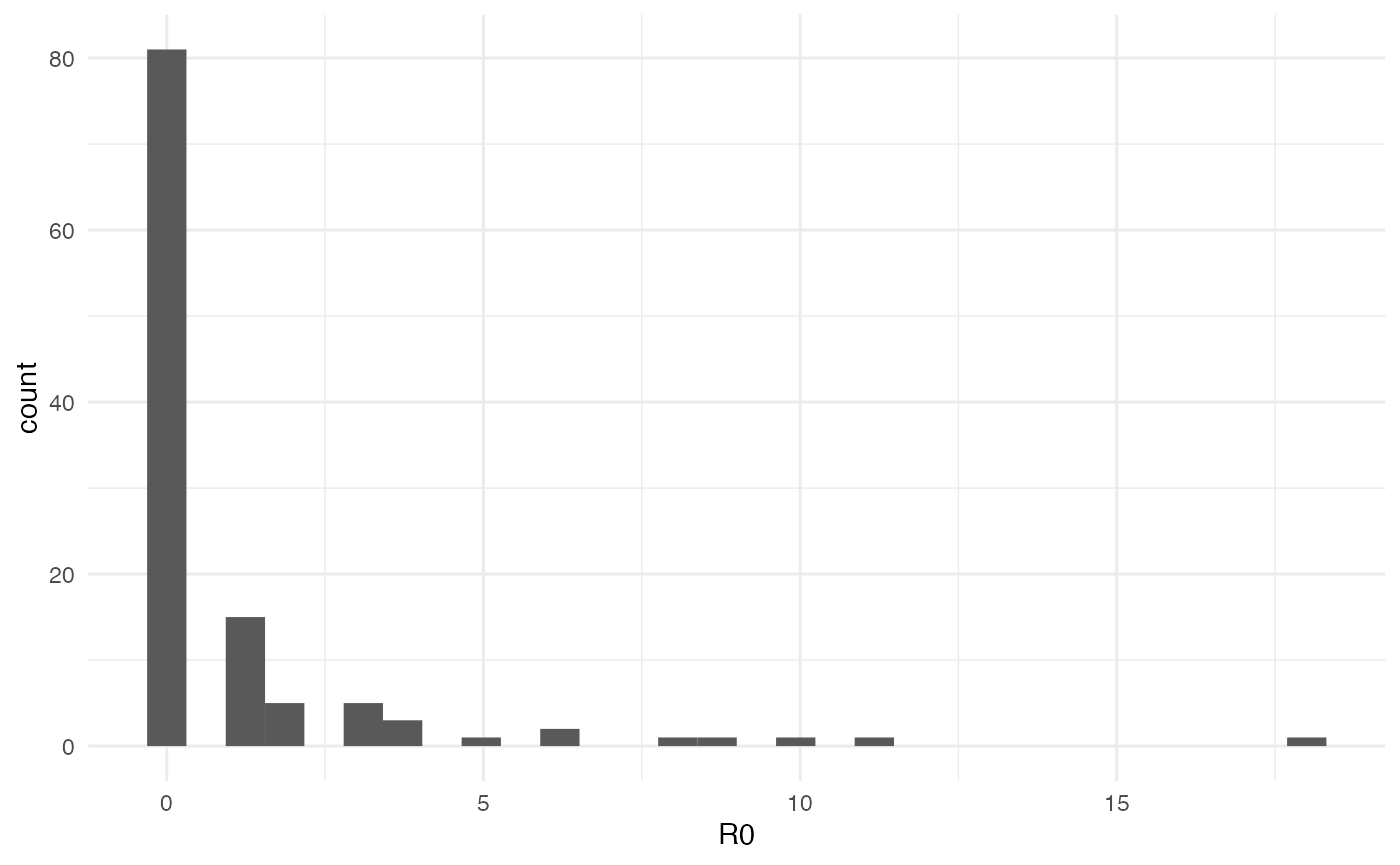
Transmission chains
Full transmission chain
The transmission chain is the main product of a nosoi
simulation. It can be extracted or visualized as such using the
table.hosts table that links hosts in time. It can also be
extracted in a form mimicking a phylogenetic tree, and visualized or
saved as such using available tools for phylogenetic trees such as ape and
tidytree.
To do so, you can use the getTransmissionTree() function,
which arguments are the nosoi simulation output and which
host type would be the tips of the tree (“A” by default, or “B” in case
a dual-host simulation).
Formally, the transmission tree is extracted as a dated phylogenetic tree where:
- each branch represents the life span of an host;
- each internal node represents a transmission event;
- each tip represents the exiting point of an host;
- the root represents the entering point of the first host.
Such a tree is binary, and has as many tips as the total number of infected hosts, and as many nodes as the number of transmission events. It spans a time going from the first entry of the first host (usually, by convention, 0), to the exiting of the last host.
It can be seen as a proxy to represent the molecular evolution of the pathogen infecting each of the hosts.
getTransmissionTree()extracts the full transmission tree. In case of a big simulated epidemic, this can take some time. It can also be very complicated to plot/visualize.
As an example, we simulate a single introduction of a single-host pathogen between 3 different states (named “A”, “B”, and “C”), with a constant exit and move probability as well as a number of contacts dependent of the location and host count in each location:
library(nosoi)
t_incub_fct <- function(x){rnorm(x,mean = 5,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
p_Move_fct <- function(t){return(0.1)}
p_Exit_fct <- function(t){return(0.05)}
proba <- function(t,p_max,t_incub){
if(t <= t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
time_contact <- function(t, current.in, host.count){
temp.val = 30 - host.count
if(temp.val <= 0) {
return(0)
}
if(temp.val >= 0) {
if(current.in=="A"){
return(round((temp.val/30)*rnorm(1, 3, 1), 0))}
if(current.in=="B"){return(0)}
if(current.in=="C"){
return(round((temp.val/30)*rnorm(1, 6, 1), 0))}
}
}
transition.matrix <- matrix(c(0,0.2,0.4,0.5,0,0.6,0.5,0.8,0),nrow = 3, ncol = 3,dimnames=list(c("A","B","C"),c("A","B","C")))
set.seed(1050)
test.nosoiA <- nosoiSim(type="single", popStructure="discrete",
length=100,
max.infected=200,
init.individuals=1,
init.structure="A",
structure.matrix=transition.matrix,
pMove=p_Move_fct,
param.pMove=NA,
diff.nContact=TRUE,
hostCount.nContact=TRUE,
nContact=time_contact,
param.nContact=NA,
pTrans = proba,
param.pTrans = list(p_max=p_max_fct,
t_incub=t_incub_fct),
pExit=p_Exit_fct,
param.pExit=NA
)The simulation runs for 46 time steps and infected 204 hosts. The
following transmission tree can thus be extracted (as a
tidytree::treedata object) and plotted using ggtree:
test.nosoiA.tree <- getTransmissionTree(test.nosoiA)
if (requireNamespace("ggtree", quietly = TRUE) &&
requireNamespace("ggplot2", quietly = TRUE)) {
library(ggtree)
library(ggplot2)
ggtree(test.nosoiA.tree, color = "gray30") +
geom_nodepoint(aes(color=state)) +
geom_tippoint(aes(color=state)) +
theme_tree2() + xlab("Time (t)") +
theme(legend.position = c(0,0.8),
legend.title = element_blank(),
legend.key = element_blank())
} else {
message("Packages 'ggtree' and 'ggplot2' are needed for plotting")
}Each color point represents the location/state, either at
transmission (a node) or a host when it exits the simulation (or the end
point of the simulation; a tip). This transmission tree is timed (time
steps of the simulation). As you can see, no transmission ever occurs at
location “B” (green); this is coherent with the chosen
nContact function, where no infectious contact occurs when
the host is in “B”.
Sampling the transmission chain
Usually, it is unlikely that the whole transmission chain (i.e. every
host) will be sampled during surveillance of an epidemic outbreak or
endemic transmission. The functions
sampleTransmissionTree() and
sampleTransmissionTreeFromExiting() can both be used to
sample hosts from this transmission chain and construct the new tree
based on the existing hosts (tips).
sampleTransmissionTree() needs the following
arguments:
-
nosoiInf, the output of simulation (nosoiSimobject) -
tree, atreedataobject created by the functiongetTransmissionTree()(see above). -
samples, adata.tableobject with the following entries:-
hosts, uniqueHost.IDof the individuals to be sampled. -
times, time at which hosts are sampled. -
labels, name of the corresponding tip in the tree
-
In the example above, we want to sample the following 20 hosts:
test.nosoiA.tree.sampled <- sampleTransmissionTree(test.nosoiA, test.nosoiA.tree, samples.data.table)As before, the tree obtained is a treedata object, and
can be plotted:
if (!requireNamespace("ggtree", quietly = TRUE)) {
ggtree(test.nosoiA.tree.sampled, color = "gray30") + geom_nodepoint(aes(color=state)) + geom_tippoint(aes(color=state)) + geom_tiplab(aes(label=host)) +
theme_tree2() + xlab("Time (t)") + theme(legend.position = c(0,0.8),
legend.title = element_blank(),
legend.key = element_blank())
}Alternatively, you can sample among exited hosts (i.e. no longer
active at the end of the simulation) using the function
sampleTransmissionTreeFromExiting(), mimicking a sampling
procedure that is destructive or cuts the hosts from the population. Be
careful however, as it does not influence the epidemiological process:
the hosts are only sampled when exiting the simulation.
sampleTransmissionTreeFromExiting() needs the following
arguments:
-
tree, atreedataobject created by the functiongetTransmissionTree()(see above). -
hosts, a vector with the hosts.ID of the exited hosts to sample.
In our example, we want to sample these samples:
test.nosoiA.tree.sampled.exiting <- sampleTransmissionTreeFromExiting(test.nosoiA.tree, sampled.hosts)As before, the tree obtained is a treedata object, and
can be plotted:
if (requireNamespace("ggtree", quietly = TRUE)) {
ggtree(test.nosoiA.tree.sampled.exiting, color = "gray30") + geom_nodepoint(aes(color=state)) + geom_tippoint(aes(color=state)) + geom_tiplab(aes(label=host)) +
theme_tree2() + xlab("Time (t)") + theme(legend.position = c(0,0.8),
legend.title = element_blank(),
legend.key = element_blank())
} else {
message("Package 'ggtree' required for plotting")
}Exporting the Transmission Tree
All the functions mentioned above produce a
tidytree::treedata object. It is a phylogenetic tree, with
nodes and tips annotated with all the characteristics of the epidemics,
including the geographical location when applicable. This format is
described in details by his developer in this ebook.
This makes it easy to export the generated data to other software for
downstream analyzes, thanks to the package treeio. For
instance, the tree can be written in a BEAST compatible format thanks to
function treeio::write.beast():
treeio::write.beast(test.nosoiA.tree.sampled.exiting)