Spread of a pathogen in a continuous space
Sebastian Lequime
2024-02-09
Source:vignettes/continuous.Rmd
continuous.RmdAside from the simple simulation set up, explored in another tutorial, where hosts are “not structured”,
nosoi can take into account a population structured either
in discrete states or in a continuous space. We focus here on a
continuous space structure (for the discrete structure, see this tutorial).
The continuous space is intended to allow the simulation to take place in a geographical context where hosts can move on a map, and may be influenced by heterogeneously distributed variables (environmental for example) on said map.
This tutorial focuses on setting up a nosoi simulation
for a pathogen which spread occurs in a continuous space.
Structure of the population
Continuous space
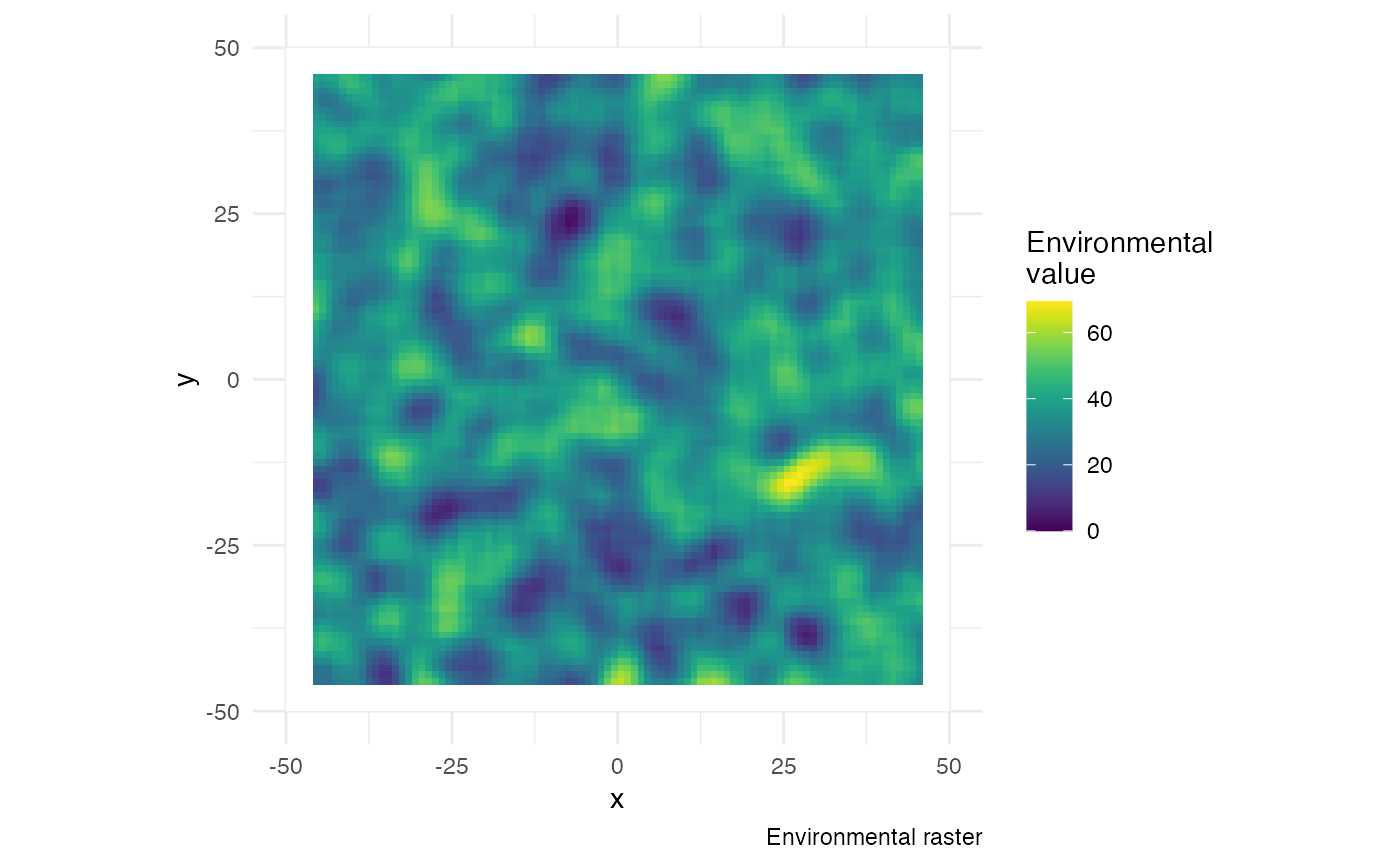
In the continuous space, the simulation takes place on a map defined
by its coordinates. This map is actually a raster, here denoted by
structure.raster, that also holds an environmental value of
interest that may influence the epidemic process. Areas were hosts
cannot move (water, inaccessible areas, etc) must be defined by the
value NA.
Here, we generate a random raster for the purpose of this tutorial, but you can use a “real” world raster and simulate your epidemic on it (see this example).
Code used to create the random raster used in this tutorial
Movement kernel
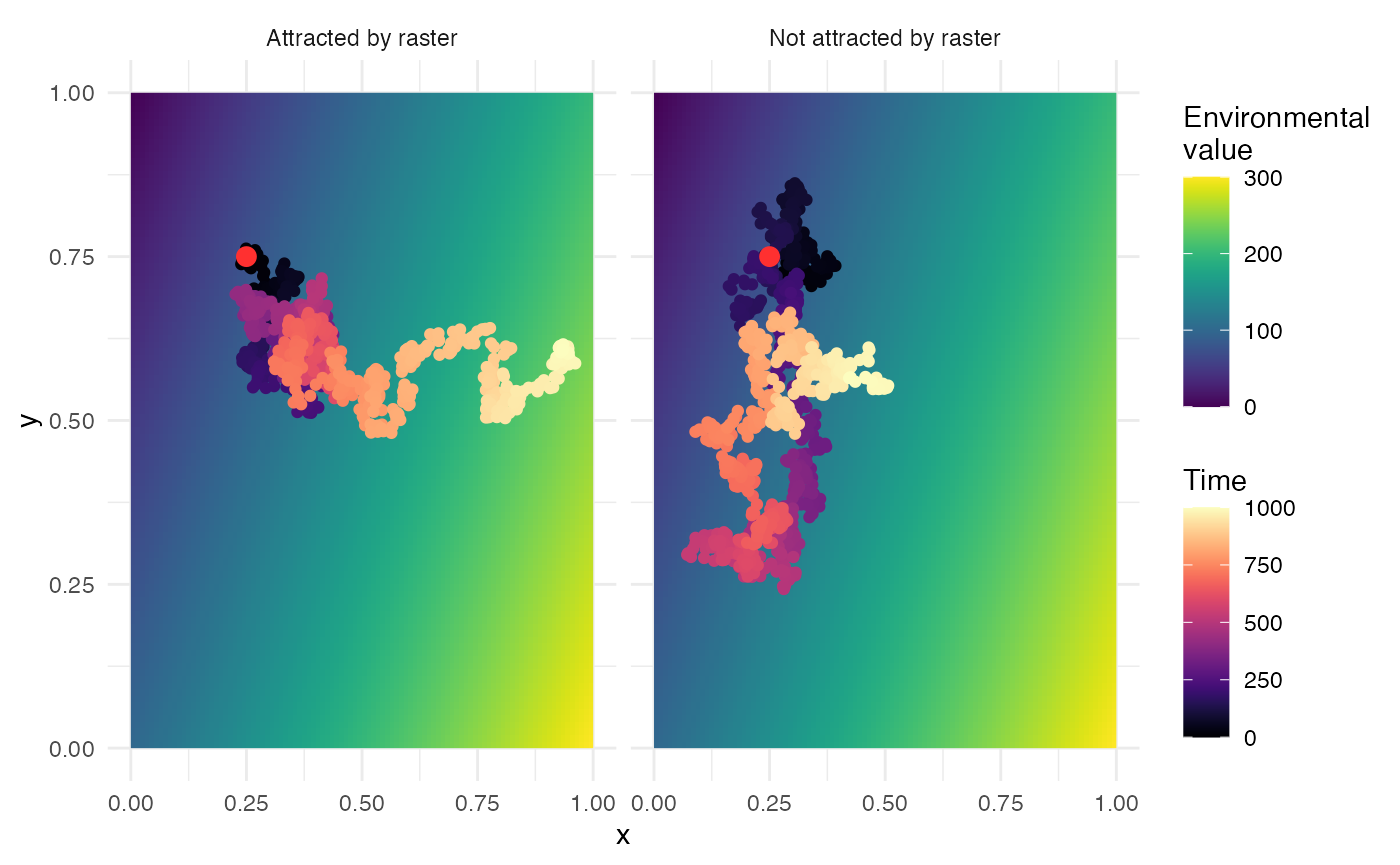
Movements in nosoi are basically described by a random
walk (Brownian motion), that can be attracted by the underlying
environmental variable (attracted.by.raster). In any case,
the movement cannot land into areas out of the raster nor into areas of
the raster with no value (NA).
The user provides a core function sdMove that gives the
standard deviation of this Brownian motion. This function, like the other core
functions, can be dependent on the time since infection
t, the absolute time of the simulation
prestime, the location (value of the environmental
variable) or individual-based parameters. This standard deviation is in
your coordinate space, so the actual distance traveled by the host may
actually vary according to your space.
In the case where hosts are attracted by the raster, hosts will tend to go to positions with high environmental values. Internally, once a new position is proposed, its environmental value is extracted and normalized (against the highest environmental value). This normalized number represents its probability of acceptance. Up to 30 draws can be made until one is accepted (by default the 30\(^{th}\) move is always accepted; this value was empirically set to allow enough exploration without being stuck in a time consuming search).
The figure below shows a movement on a simple gradient raster, with
or without an attraction (sdMove is a constant fixed at
0.01, time is 1000 units of time, maximum value of the environmental
variable is 300). The red point represents the starting position.
Setting up the simulation
The wrapper function nosoiSim takes all the arguments
that will be passed down to the simulator, in the case of this tutorial
singleContinuous (for “single host, continuous space”). We
thus start by providing the options type="single" and
popStructure="continuous" to set up the analysis:
SimulationSingle <- nosoiSim(type="single", popStructure="continuous", ...)This simulation type requires several arguments or options in order to run, namely:
length.simmax.infectedinit.individualsinit.structurestructure.raster-
pExitwithparam.pExit,timeDep.pExit,diff.pExitandhostCount.pExit -
pMovewithparam.pMove,timeDep.pMove,diff.pMoveandhostCount.pMove -
sdMovewithparam.sdMove,timeDep.sdMove,diff.sdMoveandhostCount.sdMove attracted.by.raster-
nContactwithparam.nContact,timeDep.nContact,diff.nContactandhostCount.nContact -
pTranswithparam.pTrans,timeDep.pTrans,diff.pTransandhostCount.pTrans prefix.host.Aprint.progressprint.step
All the param.* elements provide individual-level
parameters to be taken into account, while the timeDep.*
elements inform the simulator if the “absolute” simulation time should
be taken into account. The diff.* elements inform the
simulator if there is a differential probability according to the state
the host is currently in and the hostCount.* elements
inform the simulator if the number of host in each state has to be taken
into account. All parameters must be provided, although
timeDep.*, diff.* and hostCount.*
have default values set to FALSE; if you do not want to use
these options, then you do not have to explicitly provide a value.
General parameters
length.sim and max.infected are general
parameters that define the simulation:
-
length.simis the maximum number of time units (e.g. days, months, years, or another time unit of choice) during which the simulation will be run. -
max.infectedis the maximum number of individuals that can be infected during the simulation.
init.individuals and init.structure are the
“seeding parameters”:
-
init.individualsdefines the number of individuals (an integer above 1) that will start a transmission chain. Keep in mind that you will have as many transmission chains as initial individuals, which is equivalent as launching a number of independent nosoi simulations. -
init.structurespecifies the original location (as a vector of two coordinates, x and y) in the continuous space (has to be the same starting location for all starting individuals). The location provided ininit.structureshould of course be present in thestructure.raster.
Here, we will run a simulation starting with 1 individual, for a maximum of 1,000 infected individuals and a maximum time of 300 days.
SimulationSingle <- nosoiSim(type="single", popStructure="continuous",
length.sim=300, max.infected=1000, init.individuals=1, ...)Core functions
The core functions pExit, nContact,
pMove, sdMove and pTrans each
follow the same
principles to be set up.
To accommodate for different scenarios, they can be constant,
time-dependent (using the relative time since infection t
for each individual or the “absolute” time pres.time of the
simulation) or even individually parameterized, to include some
stochasticity at the individual-host level.
In any case, the provided function, like all other core functions in
nosoi, has to be expressed as a function of time
t, even if time is not used to compute the probability.
In case the function uses individual-based parameters, they must be
specified in a list of functions (called param.pExit,
param.nContact, param.pMove,
param.sdMove or param.pTrans) (see Get started). If no individual-based
parameters are used, then these lists are set to NA.
Keep in mind that
pExit,pMove, andpTranshave to return a probability (i.e. a value between 0 and 1) whilenContactshould return a natural number (positive integer or zero) andsdMovea positive number.
Several parameters, such as the time since infection, the “absolute” time of the simulation, the location (in the discrete states) and individual-based parameters can be combined within the same function.
nosoican be flexible in what it allows as parameters in your function, but a common general structure should be observed. The argument of the function should be (in that order):
- time since infection
t(compulsory);- “absolute” time
prestime(optional);- current environmental value
current.env.value(optional);- host count in state
host.count(optional);- other individual-based parameter(s), provided in
param.function.If one of the argument is not used (except
t), then you do not have to provide it and can continue with the next argument.
pExit, param.pExit,
timeDep.pExit, diff.pExit and
hostCount.pExit
-
pExitis the first required fundamental parameter and provides a daily probability for a host to leave the simulation (either cured, died, etc.). -
param.pExitis the list of functions needed to individually parameterizepExit(see Get started). The name of each function in the list has to match the name of the parameter it is sampling forpExit. -
timeDep.pExitallows forpExitto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.pExitis set toFALSE. -
diff.pExitallowspExitto differ according to the current discrete state of the host. This can be useful, for example, if one state has a higher mortality rate (or better cures!) for the infection, in that case the probability to exit the simulation is higher. By default,diff.pExitis set toFALSE. Be careful, every state should give back a result forpExit. -
hostCount.pExitallowspExitto differ according to the number of hosts currently in a state. By default,hostCount.pExitis set toFALSE. To usehostCount.pExit,diff.pExithas to be set toTRUEtoo.
pMove, param.pMove,
timeDep.pMove, diff.pMove and
hostCount.pMove
-
pMoveis the probability (per unit of time) for a host to move on the map. It should not be confused with the distance the host will travel linked tosdMove(see below). -
param.pMoveis the list of functions needed to individually parameterizepMove(see Get started). The name of each function in the list has to match the name of the parameter it is sampling forpMove. -
timeDep.pMoveallows forpMoveto be dependent of the “absolute” time of the simulation, to account, for example, for seasonality or other external time related covariates. By default,timeDep.pMoveis set toFALSE. -
diff.pMoveallowspMoveto be different according to the current environmental value of the location where the host is located. By default,diff.pMoveis set toFALSE. -
hostCount.pMoveallowspMoveto differ according to the number of hosts currently in a raster cell. By default,hostCount.pMoveis set toFALSE. To usehostCount.pMove,diff.pMovehas to be set toTRUEtoo.
sdMove, param.sdMove,
timeDep.sdMove, diff.sdMove and
hostCount.sdMove
-
sdMoverepresents the standard deviation of the Brownian motion movement when a host undergo a move (set bypMove). -
param.sdMoveis the list of function need to individually parameterizesdMove(see Get started). The name of each function in the list has to have the same name as the parameter it is sampling forsdMove. -
timeDep.sdMoveallows forsdMoveto be dependent of the “absolute” time of the simulation, to account, for example, for seasonality or other external time related covariates. By default,timeDep.sdMoveis set toFALSE. -
diff.sdMoveallowssdMoveto be different according to the current environmental value of the location where the host is located. By default,diff.sdMoveis set toFALSE. -
hostCount.sdMoveallowssdMoveto differ according to the number of hosts currently in a raster cell. By default,hostCount.sdMoveis set toFALSE. To usehostCount.sdMove,diff.sdMovehas to be set toTRUEtoo.
nContact, param.nContact,
timeDep.nContact, diff.nContact and
hostCount.nContact
-
nContactrepresents the number (expressed as a positive integer) of potentially infectious contacts an infected hosts can encounter per unit of time. At each time point, a number of contacts will be determined for each active host in the simulation. The number of contacts (i.e. the output of your function) has to be an integer and can be set to zero. -
param.nContactis the list of functions needed to individually parameterizenContact(see Get started). The name of each function in the list has to match the name of the parameter it is sampling fornContact. -
timeDep.nContactallows fornContactto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.nContactis set toFALSE. -
diff.nContactallows fornContactto differ according to the current discrete state of the host. By default,diff.nContactis set toFALSE. -
hostCount.nContactallowsnContactto differ according to the number of hosts currently in a raster cell. This can be useful to adjust the number of contact to the number of potentially susceptible hosts if the infected population is close to the maximum size of the population in a raster cell. By default,hostCount.nContactis set toFALSE. To usehostCount.nContact,diff.nContacthas to be set toTRUEtoo.
pTrans, param.pTrans,
timeDep.pTrans,diff.pTrans and
hostCount.pTrans
-
pTransis the heart of the transmission process and represents the probability of transmission over time (when a contact occurs). -
param.pTransis the list of functions needed to individually parameterizepTrans(see Get started). The name of each function in the list has to match the name of the parameter it is sampling forpTrans. -
timeDep.pTransallows forpTransto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.pTransis set toFALSE. -
diff.pTransallows forpTransto be different according to the current discrete state of the host. This can be used to account of different dynamics linked to external factors common within a state, such as temperature for example. By default,diff.pTransis set toFALSE. -
hostCount.pTransallowspTransto differ according to the number of hosts currently in a raster cell. By default,hostCount.pTransis set toFALSE. To usehostCount.pTrans,diff.pTranshas to be set toTRUEtoo.
Miscellaneous
prefix.host allows you to define the first character(s)
for the hosts’ unique ID. It will be followed by a hyphen and a unique
number. By default, prefix.host is “H” for “Host”.
print.progress allows you to have some information
printed on the screen about the simulation as it is running. It will
print something every print.step. By default,
print.progress is activated with a
print.step = 10 (you can change this frequency), but you
may want to deactivate it by setting
print.progress=FALSE.
Dual host
In the case of a dual host simulation, several parameters of the
nosoiSim will have to be specified for each host type,
designated by A and B. The wrapper function
nosoiSim will then take all the arguments that will be
passed down to the simulator, in the case of this tutorial
dualContinuous (for “dual host, continuous structure”). We
thus start by providing the options type="dual" and
popStructure="continuous" to set up the analysis:
SimulationDual <- nosoiSim(type="dual", popStructure="continuous", ...)This function takes several arguments or options to be able to run, namely:
length.simmax.infected.Amax.infected.Binit.individuals.Ainit.individuals.Binit.structure.Ainit.structure.Bstructure.raster.Astructure.raster.B-
pExit.Awithparam.pExit.A,timeDep.pExit.A,diff.pExit.AandhostCount.pExit.A -
pMove.Awithparam.pMove.A,timeDep.pMove.A,diff.pMove.AandhostCount.pMove.A -
sdMove.Awithparam.sdMove.A,timeDep.sdMove.A,diff.sdMove.AandhostCount.sdMove.A attracted.by.raster.A-
nContact.Awithparam.nContact.A,timeDep.nContact.A,diff.nContact.AandhostCount.nContact.A -
pTrans.Awithparam.pTrans.A,timeDep.pTrans.A,diff.pTrans.AandhostCount.pTrans.A prefix.host.A-
pExit.Bwithparam.pExit.B,timeDep.pExit.B,diff.pExit.BandhostCount.pExit.B -
pMove.Bwithparam.pMove.B,timeDep.pMove.B,diff.pMove.BandhostCount.pMove.B -
sdMove.Bwithparam.sdMove.B,timeDep.sdMove.B,diff.sdMove.BandhostCount.sdMove.B attracted.by.raster.B-
nContact.Bwithparam.nContact.B,timeDep.nContact.B,diff.nContact.BandhostCount.nContact.B -
pTrans.Bwithparam.pTrans.B,timeDep.pTrans.B,diff.pTrans.BandhostCount.pTrans.B prefix.host.Bprint.progressprint.step
As you can see, host-type dependent parameters are now designated by
the suffix .A or .B.
Both max.infected.A and max.infected.B have
to be provided in order to set an upper limit on the simulation size. To
initiate the simulation, you have to provide at least one starting host,
either A or B in
init.individuals.A or init.individuals.B
respectively, as well as a starting position in
init.individuals.A or init.individuals.B,
respectively. If you want to start the simulation with one host only,
then init.individuals of the other can be set to 0 and
init.structure to NA.
A major difference here is that hosts may, or not, share the same
structure.raster. However, since they exist in the same
“world”, they should be in the same coordinate system. It is also
possible to have a host that does not move. In such case,
pMove can be set to NA, as well as
sdMove. In such “non-moving” case, a raster should still be
provided, as it anchors the hosts into the simulation’s world.
Here again, all parameters must be provided for both hosts, although
timeDep, diff and hostCount have
default values set to FALSE; if you do not want to use
these options, then you do not have to explicitly provide a value. Be
careful to switch diff to TRUE if you want to
use hostCount, and remember to provide a result for each
state.
Running nosoi
Single host
We present here a very simple simulation for a single host pathogen.
pExit
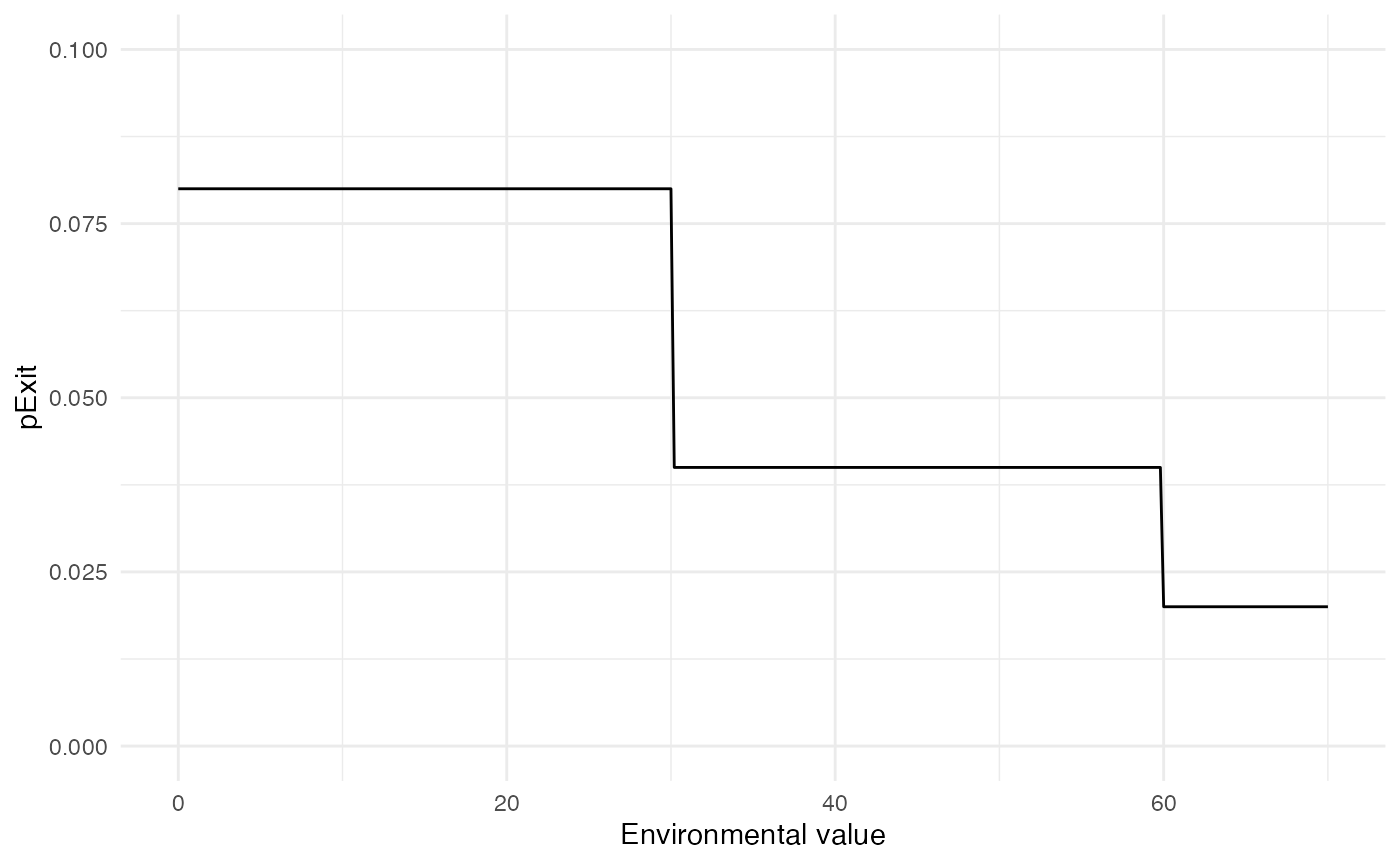
For pExit, we choose a probability that only depends on
the environmental value of the location where the host currently is. The
maximum value of our environmental raster is close to 70, and the higher
the environmental value is, the less likely the host is to die:
p_Exit_fct <- function(t, current.env.value){
if(current.env.value > 60){p=0.02}
if(current.env.value < 60 && current.env.value > 30){p=0.04}
if(current.env.value < 30){p=0.08}
return(p)
}Taken together, the value for pExit according to the
environmental value will be the following:
Remember that pExit, like the other core functions has
to be function of t, even if t is not used.
Since pExit is dependent on the location,
diff.pMove=TRUE. However, there is no use of the “absolute”
time of the simulation nor individual-based parameters, so
param.pExit=NA, and timeDep.pExit=FALSE.
pMove
We choose a constant value for pMove, namely 0.1,
i.e. an infected host has 10% chance to change its location for each
unit of time.
p_Move_fct <- function(t){return(0.1)}Remember that pMove, like the other core functions has
to be function of t, even if t is not used.
Since pMove is not dependent on the location,
diff.pMove=FALSE. Similarly, there is no use of the
“absolute” time of the simulation nor individual-based parameters, so
param.pMove=NA, and timeDep.pMove=FALSE.
sdMove
We choose a constant value for sdMove, namely 0.25.
sdMove is the standard deviation of the Brownian motion
underlying the movement in nosoi.
sd_Move_fct <- function(t){return(0.25)}Remember that sdMove, like the other core functions has
to be function of t, even if t is not used.
Since sdMove is not dependent on the location,
diff.pMove=FALSE. Similarly, there is no use of the
“absolute” time of the simulation nor individual-based parameters, so
param.sdMove=NA, and timeDep.sdMove=FALSE.
nContact
For nContact, we choose a constant function that will
draw a value in a normal distribution of mean = 0.5 and
sd = 1, round it, and take its absolute value.
The distribution of nContact looks as follows:
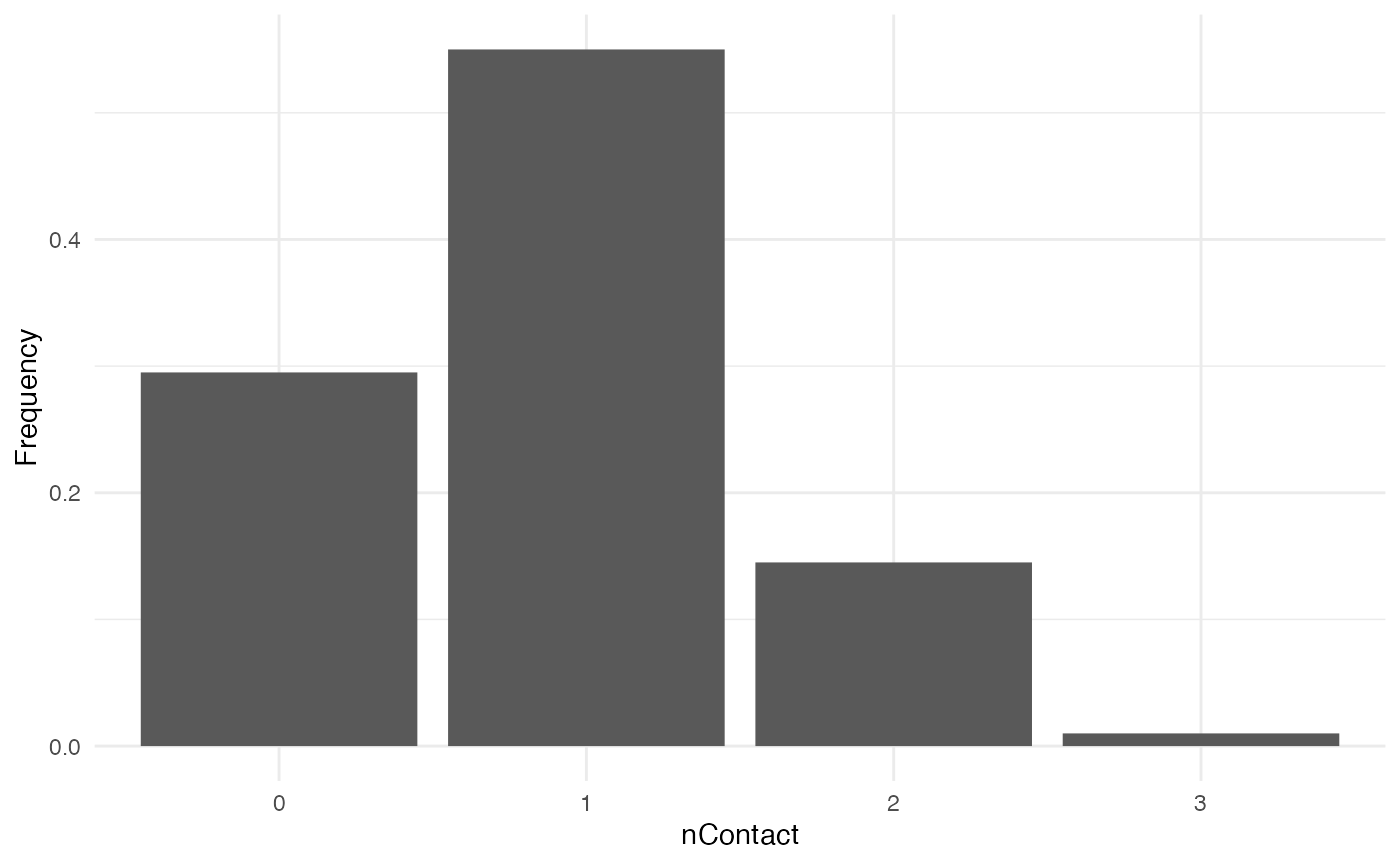
At each time and for each infected host, nContact will
be drawn anew. Remember that nContact, like the other core
functions has to be function of t, even if t
is not used. Since nContact is constant here, there is no
use of the “absolute” time of the simulation, the location of the host,
nor individual-based parameters. So param.nContact=NA,
timeDep.nContact=FALSE and
diff.nContact=FALSE.
pTrans
We choose pTrans in the form of a threshold function:
before a certain amount of time since initial infection, the host does
not transmit (incubation time, which we call t_incub), and
after that time, it will transmit with a certain (constant) probability
(which we call p_max). This function is dependent of the
time since the host’s infection t.
p_Trans_fct <- function(t, p_max, t_incub){
if(t < t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}Because each host is different (slightly different biotic and abiotic
factors), you can expect each host to exhibit differences in the
dynamics of infection, and hence the probability of transmission over
time. Thus, t_incub and p_max will be sampled
for each host individually according to a certain distribution.
t_incub will be sampled from a normal distribution of \(mean\) = 7 and \(sd\) = 1, while p_max will be
sampled from a beta distribution with shape parameters \(\alpha\) = 5 and \(\beta\) = 2:
t_incub_fct <- function(x){rnorm(x,mean = 7,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}Note that here t_incub and p_max are
functions of x and not t (they are not core
functions but individual-based parameters), and x enters
the function as the number of draws to make.
Taken together, the profile for pTrans for a subset of
200 individuals in the population will look as follows:
![]()
pTrans is not dependent on the “absolute” time of the
simulation, nor on the hosts location, so
timeDep.pTrans=FALSE and diff.pTrans=FALSE.
However, since we make use of individual-based parameters, we have to
provide a param.pTrans as a list of functions. The name of
each element within this list should have the same name that the core
function (here pTrans) uses as argument, e.g.:
Running
Once nosoiSim is set up, you can run the simulation
(here the “seed” ensures that you will get the same results as in this
tutorial).
library(nosoi)
#Raster is test.raster
#Starting position will be
start.pos <- c(0,0) # c(x,y)
#pExit
p_Exit_fct <- function(t, current.env.value){
if(current.env.value > 60){p=0.02}
if(current.env.value < 60 && current.env.value > 30){p=0.04}
if(current.env.value < 30){p=0.08}
return(p)
}
#pMove
p_Move_fct <- function(t){return(0.1)}
#sdMove
sd_Move_fct <- function(t){return(0.25)}
#nContact
n_contact_fct = function(t){abs(round(rnorm(1, 0.5, 1), 0))}
#pTrans
proba <- function(t,p_max,t_incub){
if(t <= t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
t_incub_fct <- function(x){rnorm(x,mean = 5,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
param_pTrans = list(p_max=p_max_fct,t_incub=t_incub_fct)
# Starting the simulation ------------------------------------
set.seed(846)
SimulationSingle <- nosoiSim(type="single", popStructure="continuous",
length.sim=300, max.infected=300, init.individuals=1,
init.structure=start.pos,
structure.raster=test.raster,
pExit = p_Exit_fct,
param.pExit = NA,
timeDep.pExit=FALSE,
diff.pExit=TRUE,
pMove = p_Move_fct,
param.pMove = NA,
timeDep.pMove=FALSE,
diff.pMove=FALSE,
sdMove = sd_Move_fct,
param.sdMove = NA,
timeDep.sdMove=FALSE,
diff.sdMove=FALSE,
attracted.by.raster=FALSE,
nContact=n_contact_fct,
param.nContact=NA,
timeDep.nContact=FALSE,
diff.nContact=FALSE,
pTrans = proba,
param.pTrans = list(p_max=p_max_fct,t_incub=t_incub_fct),
timeDep.pTrans=FALSE,
diff.pTrans=FALSE,
prefix.host="H",
print.progress=FALSE,
print.step=10)
#> Starting the simulation
#> Initializing ... running ...
#> done.
#> The simulation has run for 40 units of time and a total of 350 hosts have been infected.Once the simulation has finished, it reports the number of time units
for which the simulation has run (40), and the maximum number of
infected hosts (350). Note that the simulation has stopped here before
reaching length.sim as it has crossed the
max.infected threshold set at 300.
Dual host
Setting up a dual host simulation is similar to the single host version described above, but each parameter has to be provided for both hosts. Here, we choose for Host A the same parameters as the single / only host above. Host B will have sightly different parameters:
pExit.B
For pExit.B, we choose a value that depends on the
“absolute” time of the simulation, for example cyclic climatic
conditions (temperature). In that case, the function’s arguments should
be t and prestime (the “absolute” time of the
simulation), in that order:
p_Exit_fctB <- function(t,prestime){(sin(prestime/(2*pi*10))+1)/16} #for a periodic functionThe values of pExit.B across the “absolute time” of the
simulation will be the following:
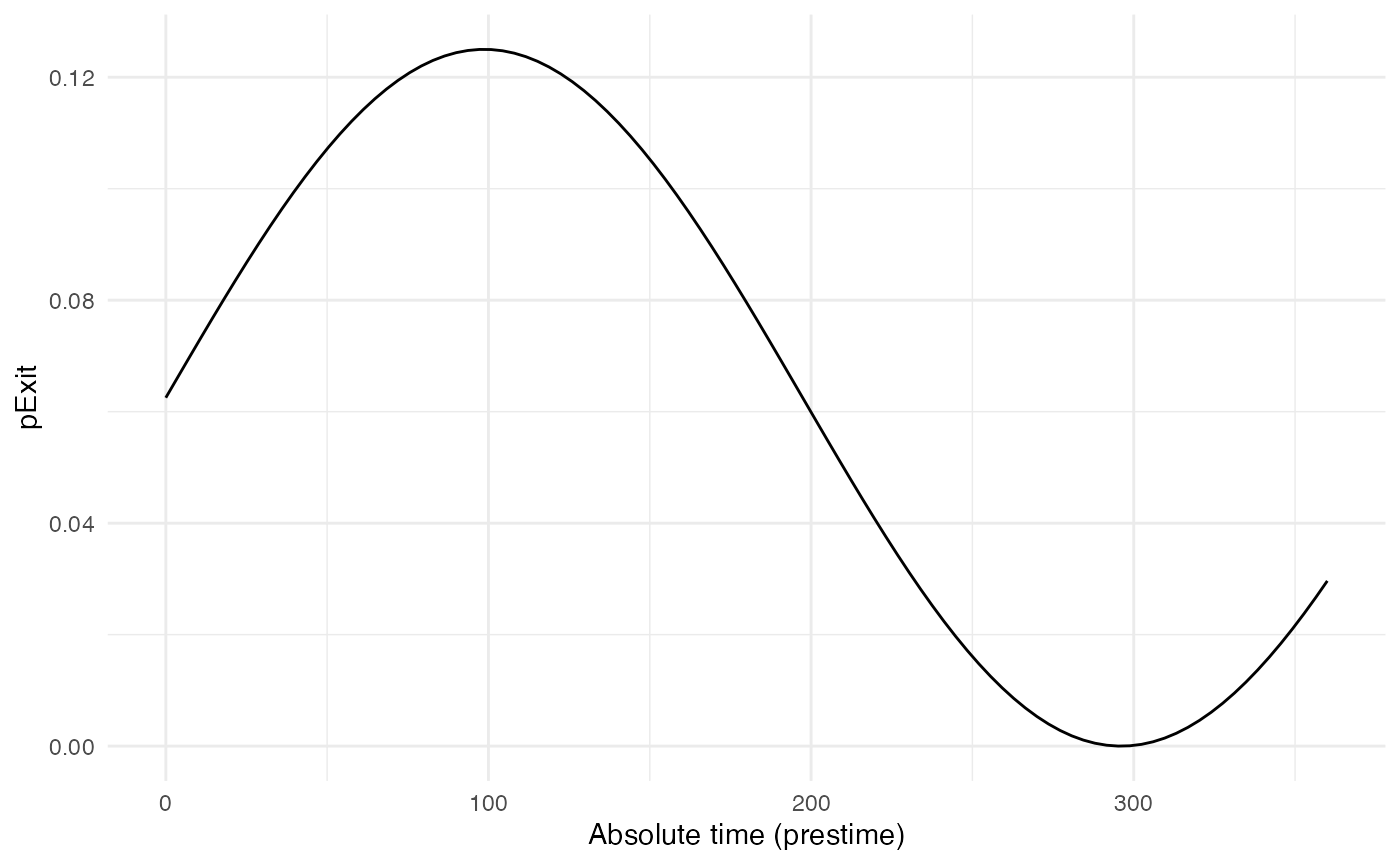
Since pExit.B is dependent on the simulation’s time, do
not forget to set timeDep.pExit.B to TRUE.
Since there are no individual-based parameters nor is there any
influence of the host’s location, param.pExit.B=NA and
diff.pExit.B=NA.
pMove.B
We assume here that the hosts B do not move. pMove.B
will hence be set to NA.
p_Move_fct.B <- NASince pMove.B is not dependent on the location,
diff.pMove.B=FALSE. Similarly, there is no use of the
“absolute” time of the simulation nor individual-based parameters, so
param.pMove.B=NA, and
timeDep.pMove.B=FALSE.
sdMove.B
Since hosts B do not move, sdMove.B is irrelevant and
will be set to NA.
sd_Move_fct.B <- NASince sdMove.B is not dependent on the location,
diff.sdMove.B=FALSE. Similarly, there is no use of the
“absolute” time of the simulation nor individual-based parameters, so
param.sdMove.B=NA, and
timeDep.sdMove.B=FALSE.
nContact.B
For nContact.B, we choose a constant function that will
sample a value from a provided list of values, with a certain
probability:
The distribution of nContact.B looks as follows:
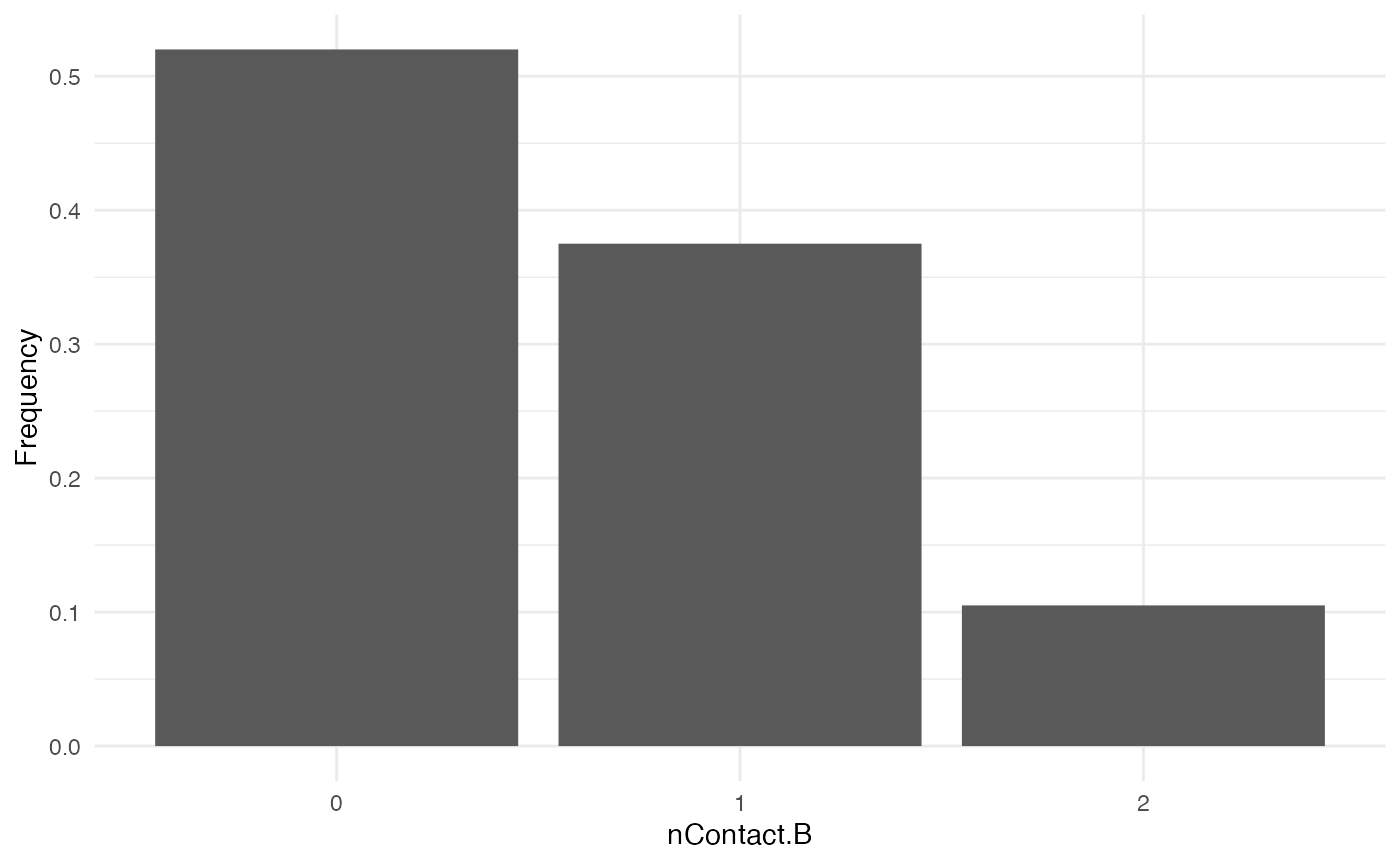
At each time and for each infected host, nContact.B will
be drawn anew. Remember that nContact.B, like the other
core functions has to be function of t, even if
t is not used. Since nContact.B is constant
here, there is no use of the “absolute” time of the simulation, the
host’s location, nor individual-based parameters. So
param.nContact.B=NA, timeDep.nContact.B=FALSE
and diff.nContact.B=FALSE.
pTrans.B
We choose pTrans.B in the form of a Gaussian function.
It will reach its maximum value at a certain time point (mean) after
initial infection and will subsequently decrease until it reaches 0.
The function dnorm used here will achieve this
objective: for each time t after infection, its return
value will reach its maximum value (< 1) at its mean (here
max.time, a parameter that will be set for each individual,
see below) and then decline back to 0. Its increase and decline is
parameterized by the standard deviation sd of the
dnorm function.
p_Trans_fct.B <- function(t, max.time){
dnorm(t, mean=max.time, sd=2)*5
}Because each host is different (slightly different biotic and abiotic
factors), you can expect each host to exhibit differences in the
dynamics of infection, and hence the probability of transmission over
time. Thus, max.time will be sampled for each host
individually according to a certain distribution. max.time
will be sampled from a normal distribution of parameters \(mean\) = 5 and \(sd\) = 1:
max.time_fct <- function(x){rnorm(x,mean = 5,sd=1)}Note again that here max.time is a function of
x and not t (i.e. not a core function but
individual-based parameters), and x enters the function as
the number of draws to make.
Taken together, the profile for pTrans for a subset of
200 individuals in the population will look as follow:
![]()
Since pTrans.B is not dependent of the “absolute” time
of the simulation nor the host’s location,
timeDep.pTrans.B=FALSE and
diff.pTrans.B=FALSE. However, since we have made use of
individual-based parameters, we have to provide a
param.pTrans as a list of functions. The names of each
element of the list should have the same name that the core function
(pTrans.B here) uses as its argument, as shown here:
Running
Once nosoiSim is set up, you can run the simulation (here the “seed” ensures that you will get the same results as in this tutorial).
library(nosoi)
#Raster is test.raster
#Starting position will be
start.pos <- c(0,0) # c(x,y)
#Host A -----------------------------------
#pExit
p_Exit_fct <- function(t, current.env.value){
if(current.env.value > 60){p=0.02}
if(current.env.value < 60 && current.env.value > 30){p=0.04}
if(current.env.value < 30){p=0.08}
return(p)
}
#pMove
p_Move_fct <- function(t){return(0.1)}
#sdMove
sd_Move_fct <- function(t){return(0.25)}
#nContact
n_contact_fct = function(t){abs(round(rnorm(1, 0.5, 1), 0))}
#pTrans
proba <- function(t,p_max,t_incub){
if(t <= t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
t_incub_fct <- function(x){rnorm(x,mean = 5,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
param_pTrans = list(p_max=p_max_fct,t_incub=t_incub_fct)
#Host B -----------------------------------
#pExit
p_Exit_fct.B <- function(t,prestime){(sin(prestime/(2*pi*10))+1)/16}
#pMove
p_Move_fct.B <- NA
#pMove
sd_Move_fct.B <- NA
#nContact
n_contact_fct.B = function(t){sample(c(0,1,2),1,prob=c(0.6,0.3,0.1))}
#pTrans
p_Trans_fct.B <- function(t, max.time){
dnorm(t, mean=max.time, sd=2)*5
}
max.time_fct <- function(x){rnorm(x,mean = 5,sd=1)}
param_pTrans.B = list(max.time=max.time_fct)
# Starting the simulation ------------------------------------
set.seed(60)
SimulationDual <- nosoiSim(type="dual", popStructure="continuous",
length.sim=300,
max.infected.A=100,
max.infected.B=200,
init.individuals.A=1,
init.individuals.B=0,
init.structure.A=start.pos,
init.structure.B=NA,
structure.raster.A=test.raster,
structure.raster.B=test.raster,
pExit.A = p_Exit_fct,
param.pExit.A = NA,
timeDep.pExit.A=FALSE,
diff.pExit.A=TRUE,
pMove.A = p_Move_fct,
param.pMove.A = NA,
timeDep.pMove.A=FALSE,
diff.pMove.A=FALSE,
sdMove.A = sd_Move_fct,
param.sdMove.A = NA,
timeDep.sdMove.A=FALSE,
diff.sdMove.A=FALSE,
attracted.by.raster.A=FALSE,
nContact.A=n_contact_fct,
param.nContact.A=NA,
timeDep.nContact.A=FALSE,
diff.nContact.A=FALSE,
pTrans.A = proba,
param.pTrans.A = list(p_max=p_max_fct,t_incub=t_incub_fct),
timeDep.pTrans.A=FALSE,
diff.pTrans.A=FALSE,
prefix.host.A="H",
pExit.B = p_Exit_fct.B,
param.pExit.B = NA,
timeDep.pExit.B=TRUE,
diff.pExit.B=FALSE,
pMove.B = p_Move_fct.B,
param.pMove.B = NA,
timeDep.pMove.B=FALSE,
diff.pMove.B=FALSE,
sdMove.B = sd_Move_fct.B,
param.sdMove.B = NA,
timeDep.sdMove.B=FALSE,
diff.sdMove.B=FALSE,
attracted.by.raster.B=FALSE,
nContact.B=n_contact_fct.B,
param.nContact.B=NA,
timeDep.nContact.B=FALSE,
diff.nContact.B=FALSE,
pTrans.B = p_Trans_fct.B,
param.pTrans.B = param_pTrans.B,
timeDep.pTrans.B=FALSE,
diff.pTrans.B=FALSE,
prefix.host.B="V",
print.progress=FALSE)
#> Starting the simulation
#> Initializing ... running ...
#> done.
#> The simulation has run for 33 units of time and a total of 102 (A) and 137 (B) hosts have been infected.Once the simulation has finished, it reports the number of time units
for which the simulation has run (33), and the maximum number of
infected hosts A (102) and hosts B (137). Note that the simulation has
stopped here before reaching length.sim as it has crossed
the max.infected.A threshold set at 100.
Going further
To analyze and visualize your nosoi simulation output,
you can have a look on this
page.
A practical example using a dual host type of simulation with population structure in a continuous space is also available: