Spread of a pathogen in a homogeneous population
Sebastian Lequime
2024-02-09
Source:vignettes/none.Rmd
none.Rmdnosoi can accommodate a wide range of epidemiological
transmission scenarios.
It
hence relies on many parameters, that need to be set properly for the
right scenario to be simulated. This tutorial aims to illustrate how to
set up a nosoi simulation for a “simple” case: a pathogen
being transmitted within a population without structure. We will present
two cases, first for a single-host, and then a dual-host pathogen.
Setting up the simulation
The wrapper function nosoiSim takes all the arguments
that will be passed down to the simulator, in the case of this tutorial
singleNone (for “single host, no structure”). We thus start
by providing the options type="single" and
popStructure="none" to set up the analysis:
SimulationSingle <- nosoiSim(type="single", popStructure="none", ...)This simulation type requires several arguments or options in order to run, namely:
length.simmax.infectedinit.individuals-
pExitwithparam.pExitandtimeDep.pExit -
nContactwithparam.nContactandtimeDep.nContact -
pTranswithparam.pTransandtimeDep.pTrans prefix.hostprogress.barprint.step
All the param.* elements provide individual-level
parameters to be taken into account, while the timeDep.*
elements inform the simulator if the “absolute” simulation time should
be taken into account.
General parameters
length.sim, max.infected and
init.individuals are general parameters that define the
simulation:
-
length.simis the maximum number of time units (e.g. days, months, years, or another time unit of choice) during which the simulation will be run. -
max.infectedis the maximum number of individuals that can be infected during the simulation. -
init.individualsdefines the number of individuals (an integer above 1) that will start a transmission chain (there will be as many transmission chains as initial individuals that “seed” the epidemic process).
Here, we will run a simulation starting with 1 individual, for a maximum of 1,000 infected individuals and a maximum time of 300 days.
SimulationSingle <- nosoiSim(type="single", popStructure="none",
length.sim=300, max.infected=1000, init.individuals=1, ...)Core functions
The core functions pExit, nContact and
pTrans each follow the same principles to be
set up.
To accommodate for different scenarios, they can be constant,
time-dependent (using the relative time since infection t
for each individual or the “absolute” time pres.time of the
simulation) or even individually parameterized, to include some
stochasticity at the individual-host level.
In any case, the provided function, like all other core functions in
nosoi, has to be expressed as a function of time
t, even if time is not used to compute the probability.
In case the function uses individual-based parameters, they must be
specified in a list of functions (called param.pExit,
param.nContact or param.pTrans) (see Get started). If no individual-based
parameters are used, then these lists are set to NA.
Keep in mind that
pExitandpTranshave to return a probability (i.e. a number between 0 and 1) whilenContactshould return a natural number (positive integer or zero).
Several parameters, such as the time since infection, the “absolute” time of the simulation and individual-based parameters can be combined within the same function.
In any case, time since infection and “absolute” time should ALWAYS be designated by
tandprestimerespectively. They also have to be used in the order: (1)t; (2)prestimeand (3) individual-based parameters. This is necessary for the function to be properly parsed bynosoi.
pExit, param.pExit and
timeDep.pExit
-
pExitis the first required fundamental parameter and provides a daily probability for a host to leave the simulation (either cured, died, etc.). -
param.pExitis the list of functions needed to individually parameterizepExit(see Get started). The name of each function in the list has to match the name of the parameter it is sampling forpExit. -
timeDep.pExitallows forpExitto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.pExitis set toFALSE.
nContact, param.nContact and
timeDep.nContact
-
nContactrepresents the number (expressed as a positive integer) of potentially infectious contacts an infected hosts can encounter per unit of time. At each time point, a number of contacts will be determined for each active host in the simulation. The number of contacts (i.e. the output of your function) has to be an integer and can be set to zero. -
param.nContactis the list of functions needed to individually parameterizenContact(see Get started). The name of each function in the list has to match the name of the parameter it is sampling fornContact. -
timeDep.nContactallows fornContactto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.nContactis set toFALSE.
pTrans, param.pTrans and
timeDep.pTrans
-
pTransis the heart of the transmission process and represents the probability of transmission over time (when a contact occurs). -
param.pTransis the list of functions needed to individually parameterizepTrans(see Get started). The name of each function in the list has to match the name of the parameter it is sampling forpTrans. -
timeDep.pTransallows forpTransto be dependent on the “absolute” time of the simulation, to account - for example - for seasonality or other external time-related covariates. By default,timeDep.pTransis set toFALSE.
Miscellaneous
prefix.host allows you to define the first character(s)
for the hosts’ unique ID. It will be followed by a hyphen and a unique
number. By default, prefix.host is “H” for “Host”.
print.progress allows you to have some information
printed on the screen about the simulation as it is running. It will
print something every print.step. By default,
print.progress is activated with a
print.step = 10 (you can change this frequency), but you
may want to deactivate it by setting
print.progress=FALSE.
Dual host
In the case of a dual host simulation, several parameters of the
nosoiSim will have to be specified for each host type,
designated by A and B. The wrapper function
nosoiSim will then take all the arguments that will be
passed down to the simulator, in the case of this tutorial
dualNone (for “dual host, no structure”). We thus start by
providing the options type="dual" and
popStructure="none" to set up the analysis:
SimulationDual <- nosoiSim(type="dual", popStructure="none", ...)As with singleNone, this function takes several
arguments or options to be able to run, namely:
length.simmax.infected.Amax.infected.Binit.individuals.Ainit.individuals.B-
pExit.Awithparam.pExit.AandtimeDep.pExit.A -
nContact.Awithparam.nContact.AandtimeDep.nContact.A -
pTrans.Awithparam.pTrans.AandtimeDep.pTrans.A prefix.host.A-
pExit.Bwithparam.pExit.BandtimeDep.pExit.B -
nContact.Bwithparam.nContact.BandtimeDep.nContact.B -
pTrans.Bwithparam.pTrans.BandtimeDep.pTrans.B prefix.host.Bprint.progressprint.step
As you can see, host-type dependent parameters are now designated by
the suffix .A or .B.
Both max.infected.A and max.infected.B have
to be provided to set an upper limit on the simulation size. To initiate
the simulation, you have to provide at least one starting host, either
A or B in init.individuals.A or
init.individuals.B respectively. If you want to start the
simulation with one host only, then the init.individuals of
the other can be set to 0.
Running nosoi
Single host
We present here a very simple simulation for a single host pathogen.
pExit
For pExit, we choose a constant value, namely 0.08,
i.e. an infected host has 8% chance to leave the simulation at each unit
of time:
p_Exit_fct <- function(t){return(0.08)}Remember that pExit, like the other core functions has
to be function of t, even if t is not used.
Since pExit is constant here, there is no use for the
“absolute” time of the simulation nor for the individual-based
parameters. So param.pExit=NA, and
timeDep.pExit=FALSE.
nContact
For nContact, we choose a constant function that will
draw a value from a normal distribution with mean = 0.5 and
sd = 1, round it, and take its absolute value:
The distribution of nContact looks as follows:
#>
#> Attaching package: 'dplyr'
#> The following objects are masked from 'package:stats':
#>
#> filter, lag
#> The following objects are masked from 'package:base':
#>
#> intersect, setdiff, setequal, union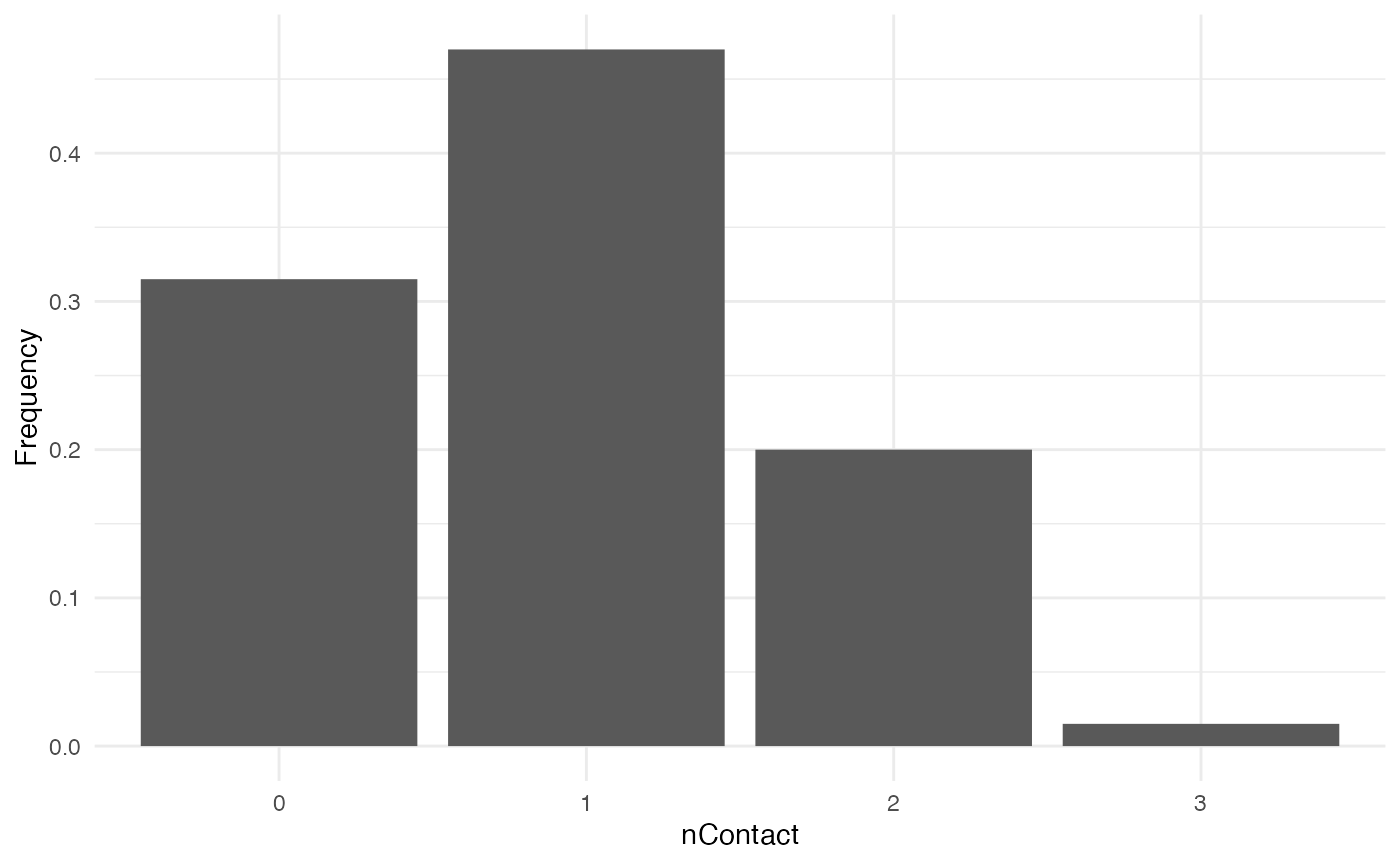
At each time step and for each infected host, nContact
will be drawn anew. Remember that nContact, like the other
core functions has to be function of t, even if
t is not used. Since nContact is constant
here, there is no use for the “absolute” time of the simulation nor for
the individual-based parameters. So param.nContact=NA, and
timeDep.nContact=FALSE.
pTrans
We choose pTrans in the form of a threshold function:
before a certain amount of time since initial infection, the host does
not transmit (incubation time, which we call t_incub), and
after that time it will transmit with a certain (constant) probability
(which we call p_max). This function is dependent on the
time since the host’s infection t:
p_Trans_fct <- function(t, p_max, t_incub){
if(t < t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}Because each host is different (slightly different biotic and abiotic
factors), you can expect each host to exhibit differences in the
dynamics of infection, and hence the probability of transmission over
time. Thus, t_incub and p_max will be sampled
for each host individually according to a certain distribution.
t_incub will be sampled from a normal distribution of \(mean\) = 7 and \(sd\) = 1, while p_max will be
sampled from a beta distribution with shape parameters \(\alpha\) = 5 and \(\beta\) = 2:
t_incub_fct <- function(x){rnorm(x,mean = 7,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}Note that here t_incub and p_max are
functions of x and not t (they are not core
functions but individual-based parameters), and x enters
the function as the number of draws to make.
Taken together, the profile for pTrans for a subset of
200 individuals in the population will look as follows:
![]()
pTrans is not dependent on the “absolute” time of the
simulation, so timeDep.pTrans=FALSE. However, since we make
use of individual-based parameters, we have to provide a
param.pTrans as a list of functions. The name of each
element within this list should have the same name that the core
function (here pTrans) uses as argument, e.g.:
Running
Once nosoiSim is set up, you can run the simulation
(here the “seed” ensures that you will obtain the same results as in
this tutorial):
library(nosoi)
#> Loading required package: data.table
#>
#> Attaching package: 'data.table'
#> The following objects are masked from 'package:dplyr':
#>
#> between, first, last
#pExit
p_Exit_fct <- function(t){return(0.08)}
#nContact
n_contact_fct = function(t){abs(round(rnorm(1, 0.5, 1), 0))}
#pTrans
p_Trans_fct <- function(t,p_max,t_incub){
if(t < t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
t_incub_fct <- function(x){rnorm(x,mean = 7,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
param_pTrans = list(p_max=p_max_fct,t_incub=t_incub_fct)
# Starting the simulation ------------------------------------
set.seed(805)
SimulationSingle <- nosoiSim(type="single", popStructure="none",
length.sim=100, max.infected=100, init.individuals=1,
nContact=n_contact_fct,
param.nContact=NA,
timeDep.nContact=FALSE,
pExit = p_Exit_fct,
param.pExit=NA,
timeDep.pExit=FALSE,
pTrans = p_Trans_fct,
param.pTrans = param_pTrans,
timeDep.pTrans=FALSE,
prefix.host="H",
print.progress=FALSE)
#> Starting the simulation
#> Initializing ...
#> running ...
#> done.
#> The simulation has run for 40 units of time and a total of 111 hosts have been infected.Once the simulation has finished, it reports the number of time units
for which the simulation has run (40), and the maximum number of
infected hosts (111). Note that the simulation has stopped here before
reaching length.sim as it has crossed the
max.infected threshold set at 100.
Dual host
Setting up a dual host simulation is similar to the single host version described above, but each parameter has to be provided for both hosts. Here, we choose for Host A the same parameters as the single / only host above. Host B will have sightly different parameters:
pExit.B
For pExit.B, we choose a value that depends on the
“absolute” time of the simulation, for example cyclic climatic
conditions (temperature). In that case, the function’s arguments should
be t and prestime (the “absolute” time of the
simulation), in that order:
p_Exit_fctB <- function(t,prestime){(sin(prestime/(2*pi*10))+1)/16} #for a periodic functionThe values of pExit.B across the “absolute time” of the
simulation will be the following:
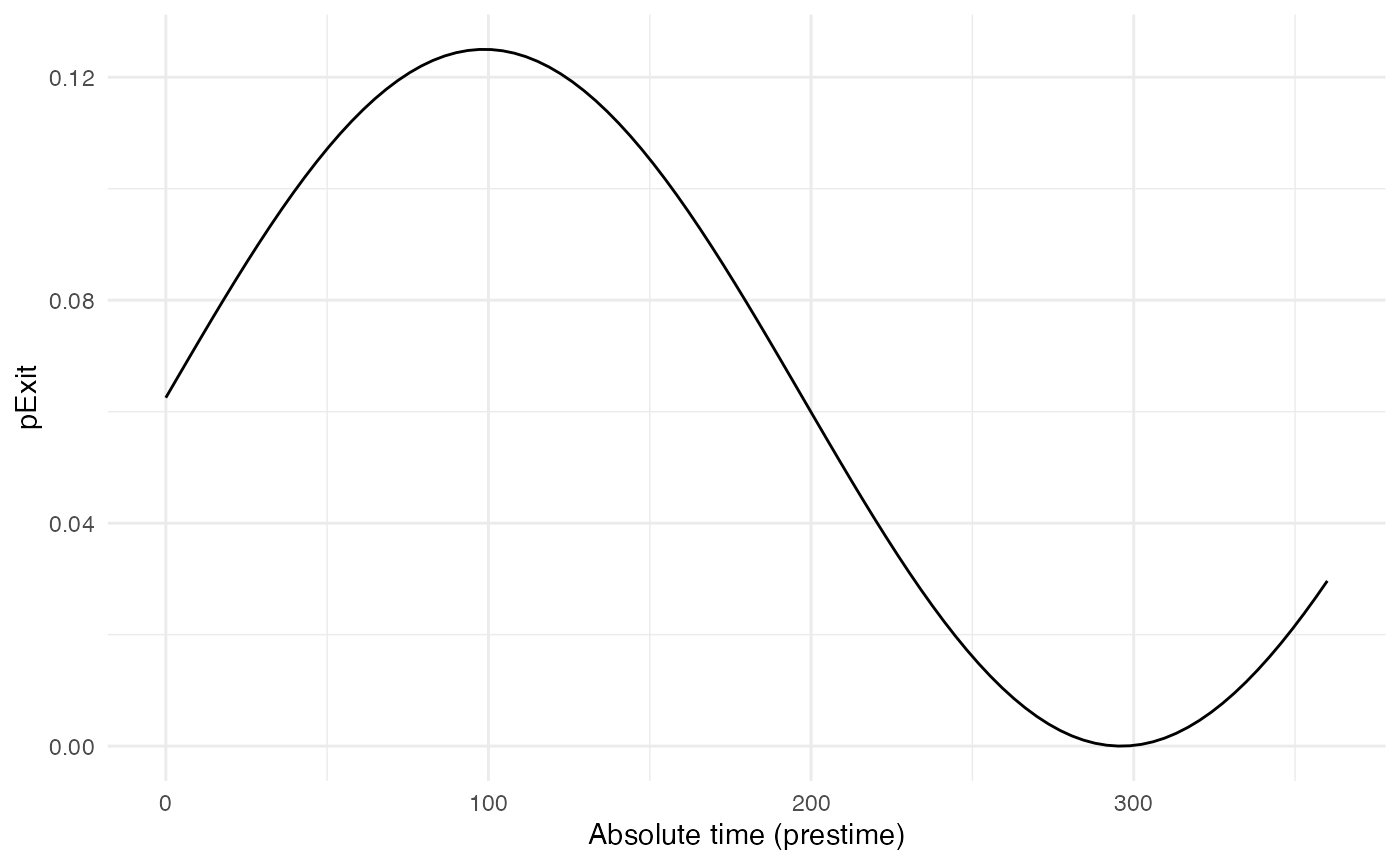
Since pExit.B is dependent on the simulation’s absolute
time, do not forget to set timeDep.pExit.B to
TRUE. Since there are no individual-based parameters,
param.pExit.B=NA.
nContact.B
For nContact.B, we choose a constant function that will
sample a value out of a provided range of possible values, each with a
certain probability:
The distribution of nContact.B looks as follows:
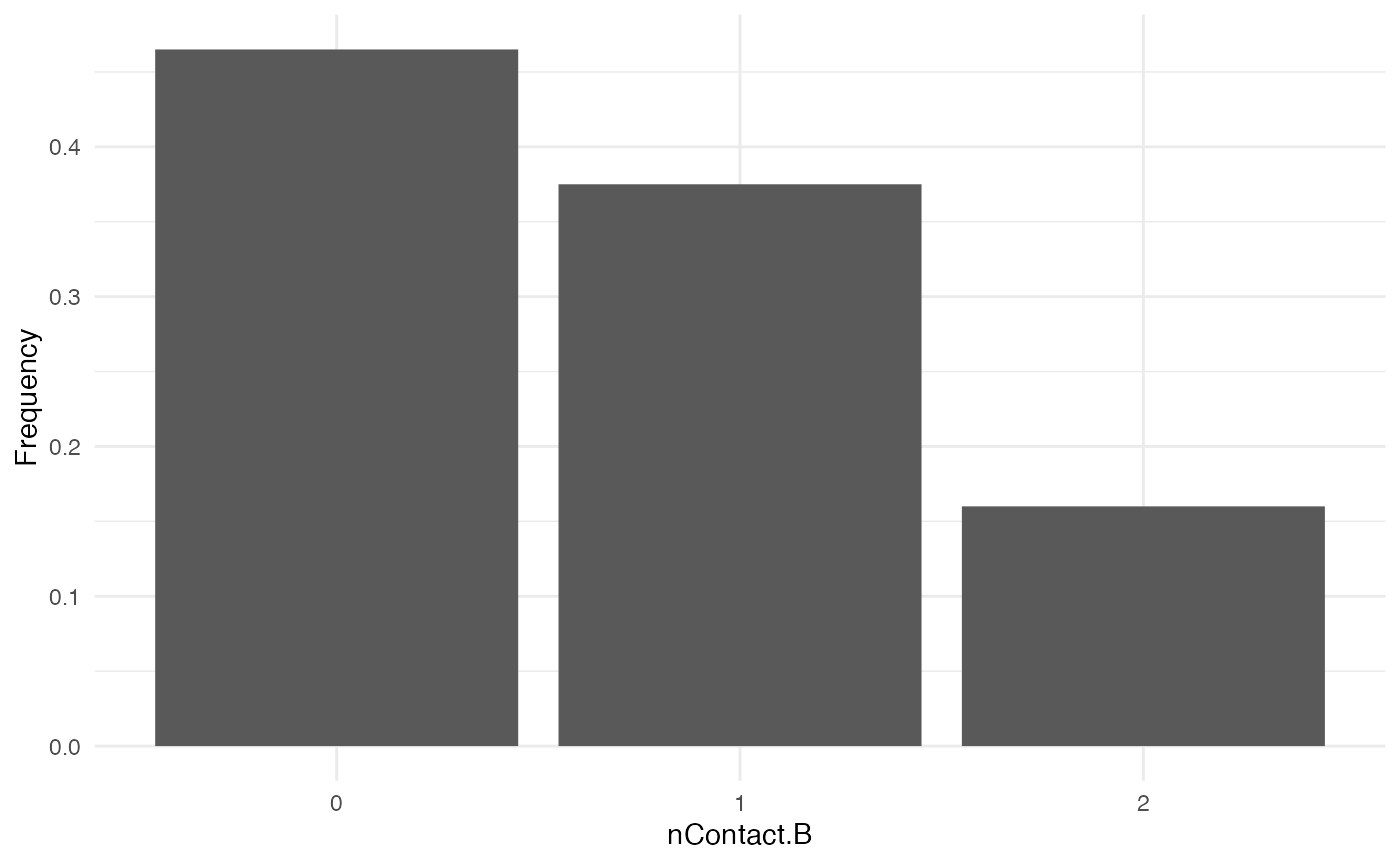
At each time and for each infected host, nContact.B will
be drawn anew. Remember that nContact.B, like the other
core functions has to be function of t, even if
t is not used. Since nContact.B is constant
here, there is no use for the “absolute” time of the simulation nor for
the individual-based parameters. So param.nContact.B=NA,
and timeDep.nContact.B=FALSE.
pTrans.B
We choose pTrans.B in the form of a Gaussian function.
It will reach its maximum value at a certain time point (mean) after
initial infection and will subsequently decrease until it reaches 0:
p_Trans_fct.B <- function(t, max.time){
dnorm(t, mean=max.time, sd=2)*5
}Because each host is different (slightly different biotic and abiotic
factors), you can expect each host to exhibit differences in the
dynamics of infection, and hence the probability of transmission over
time. Thus, max.time will be sampled for each host
individually according to a certain distribution. max.time
will be sampled from a normal distribution of parameters \(mean\) = 5 and \(sd\) = 1:
max.time_fct <- function(x){rnorm(x,mean = 5,sd=1)}Note again that here max.time is a function of
x and not t (not a core function but
individual-based parameters), and x enters the function as
the number of draws to make.
Taken together, the profile for pTrans for a subset of 200 individuals in the population will look as follows:
![]()
Since pTrans.B is not dependent on the “absolute” time
of the simulation, timeDep.pTrans.B=FALSE. However, since
we make use of individual-based parameters, we have to provide a
param.pTrans as a list of functions. The name of each
element of the list should have the same name as the core function (here
pTrans.B) uses as argument, as shown here:
Running
Once nosoiSim is set up, you can run the simulation
(here the “seed” ensures that you will obtain the same results as in
this tutorial):
library(nosoi)
#HostA ------------------------------------
#pExit
p_Exit_fct.A <- function(t){return(0.08)}
#nContact
n_contact_fct.A = function(t){abs(round(rnorm(1, 0.5, 1), 0))}
#pTrans
p_Trans_fct.A <- function(t,p_max,t_incub){
if(t < t_incub){p=0}
if(t >= t_incub){p=p_max}
return(p)
}
t_incub_fct <- function(x){rnorm(x,mean = 7,sd=1)}
p_max_fct <- function(x){rbeta(x,shape1 = 5,shape2=2)}
param_pTrans.A = list(p_max=p_max_fct,t_incub=t_incub_fct)
#Host B ------------------------------------
#pExit
p_Exit_fct.B <- function(t,prestime){(sin(prestime/(2*pi*10))+1)/16}
#nContact
n_contact_fct.B = function(t){sample(c(0,1,2),1,prob=c(0.6,0.3,0.1))}
#pTrans
p_Trans_fct.B <- function(t, max.time){
dnorm(t, mean=max.time, sd=2)*5
}
max.time_fct <- function(x){rnorm(x,mean = 5,sd=1)}
param_pTrans.B = list(max.time=max.time_fct)
# Starting the simulation ------------------------------------
set.seed(606)
SimulationDual <- nosoiSim(type="dual", popStructure="none",
length.sim=100,
max.infected.A=100,
max.infected.B=100,
init.individuals.A=1,
init.individuals.B=0,
nContact.A=n_contact_fct.A,
param.nContact.A=NA,
timeDep.nContact.A=FALSE,
pExit.A=p_Exit_fct.A,
param.pExit.A=NA,
timeDep.pExit.A=FALSE,
pTrans.A=p_Trans_fct.A,
param.pTrans.A=param_pTrans.A,
timeDep.pTrans.A=FALSE,
prefix.host.A="H",
nContact.B=n_contact_fct.B,
param.nContact.B=NA,
timeDep.nContact.B=FALSE,
pExit.B=p_Exit_fct.B,
param.pExit.B=NA,
timeDep.pExit.B=TRUE,
pTrans.B=p_Trans_fct.B,
param.pTrans.B=param_pTrans.B,
timeDep.pTrans.B=FALSE,
prefix.host.B="V",
print.progress=FALSE)
#> Starting the simulation
#> Initializing ... running ...
#> done.
#> The simulation has run for 43 units of time and a total of 101 (A) and 92 (B) hosts have been infected.Once the simulation has finished, it reports the number of time units
for which the simulation has run (43), and the maximum number of
infected hosts A (101) and hosts B (92). Note that the simulation has
stopped here before reaching length.sim as it has crossed
the max.infected.A threshold set at 100.
Going further
To analyze and visualize your nosoi simulation output,
you can have a look on this
page.
You may also want to compose a more complex model by adding some structure (e.g. geography) to your simulation. Two tutorials can guide you on how to set up such structured scenarios:
- Spread of a pathogen in a structured (discrete) population of hosts.
- Spread of a pathogen in a structure (continuous) population of hosts.
A practical example using a dual host type of simulation without population structure is also available: